
DNA Damage Detection and its Impact on the Cell Cycle
Monika Kurpas, Katarzyna Jonak and Krzysztof Puszynski
Faculty of Automatic Control, Electronics and Computer Science, Silesian University of Technology,
Akademicka 16, 44-102 Gliwice, Poland
1 STAGE OF THE RESEARCH
1.1 Biological Background
Thousands of DNA lesions are formed daily in each
cell of the human body. They can be induced either
endogenously, as well as exogenously - by physical
and chemical agents from outside of the body. There
are several types of DNA damage, from small chem-
ical modifications of single-stranded DNA through
the photoproducts and adducts caused by UV irradia-
tion, to the potentially most dangerous double-strand
breaks. The existence of such a large number of ab-
normalities in many cells may cause the death of the
body after a very short time. During evolution num-
ber of mechanisms that protect cell from damages
evolved to prevent cell death and lesions transforma-
tion to future generations. There are several path-
ways of DNA repair depending on nature of dam-
age, their review can be found in (Ciccia and Elledge,
2010). For all of these complex mechanisms of re-
Figure 1: DNA damage detection and signal amplification.
pair, it is necessary to detect DNA damage just af-
ter it arises and spread the information about it to the
proper regulatory units. This process takes place in a
manner specific to the type of lesion. ATR (ataxia
telangiectasia mutated and Rad3-related) module is
activated by presence of single stranded DNA areas
in the cell, which are caused by resection of various
types of lesions or by stalled replication forks. Dou-
ble strand breaks (DSBs) are detected indirectly by
ataxia telangiectasia mutated (ATM). Stages of DNA
damage detection by ATR and ATM and its further
amplification are presented in fig. 1.
In case of ATR subpathway, the signal strength
of the checkpoint cascade is dependent on the
length of RPA-ssDNA regions and possibility of ATR
molecules located closely to each other. Other ATR
phosphorylation targets are RPA subunits (RPA70 and
RPA32), ATRIP, TopBP1, Chk2, p53 and histone
H2AX. Double strand breaks are detected by repair
complexes like MRN complex. MRN binds to DNA
damage site and recruits ATM kinase, that after its
autophosphorylation interacts with several protein in
pathway (Chk1, Chk2, p53, Mdm, CREB, Wip1) and
leads to p53 stabilization.
1.2 ATR-p53 Model
Our existing stochastic mathematical model of ATR
signaling pathway is based on the Haseltine-Rawlings
postulate (Haseltine and Rawlings, 2002) and is an
extension of our previous model of the p53 signaling
pathway (Puszynski et al., 2008). The model is
activated by UV irradiation which results in SSBs
lesions occurrence. The output of the model is the
level of p53 protein which determines cell fate: DNA
damage repair or cell apoptosis. Spontaneous DNA
damage formation implemented in presented model
results in basic ATR pathway activation. The core
of the model are states of ATR: inactive protein, its
phosphorylated state, and fully activated form. In this
model, there are two feedback loops: positive, with
the participation of PTEN protein, and negative, con-
taining MDM - p53 suppressor (fig. 2). Details about
67
Kurpas M., Jonak K. and Puszy
´
nski K..
DNA Damage Detection and its Impact on the Cell Cycle.
Copyright
c
2014 SCITEPRESS (Science and Technology Publications, Lda.)

Figure 2: DNA damage detection model vizualization. Solid lines represent change of protein form; dashed lines describe the
interactions that occur in the path. Components of ATR module are colored in blue.
p53 signaling pathway are available in (Puszynski
et al., 2008). The model distinguish the nucleus and
cytoplasm. It was assumed that each gene has two
copies. None of them can be active, one of them or
both can be active. For some proteins production
and degradation was not modeled directly assuming
that they are equal and protein only change the form
(from active to inactive and vice versa). In presented
model a simplified DNA repair was implementing
depending on the number of p53 tetramers, repair rate
and the amount of repair complexes, which is limited.
Apoptosis condition is recognized as a permanently
elevated level of the p53 protein (over 6 hours). Then
the cell dies and all of its elements are degraded, thus
further protein levels and the number of lesions are
not taken into account.
1.2.1 Simulation Analysis of ATR Module
In the simulation analysis we examined cell response
to different doses of radiation, we set the threshold
of detection and apoptosis, as well as showed sponta-
neous activation of the ATR/p53 pathway. Determin-
istic and stochastic (for 100 cells) experiments were
performed. At t=24 hours after start, simulated cells
were irradiated by a specific dose of UVC, and then
observed over the next 48 hours. The correctness of
the model was verified based on the results of biolog-
ical experiments from the literature.
3A. Basic Activation
According to (Kohn, 2002) in every cell of the human
body in a day are formed approximately 55 000
single-strand breaks, which are responsible for base
activation of the path (fig. 3A).
3B. Damage Detection Threshold
We examined response of the model (fig. 3B) to ra-
diation dose causing one single strand break (0.0665
mJ/m
2
).
4A. Apoptotic Death Threshold
Based on the observation of simulation results, the
dose of 18 J/m
2
in which more than half of the cell
becomes apoptotic cells was taken as the threshold
for apoptosis (fig. 4A). . For comparison, the dose
17 J/m
2
causing death of 44/100 cells. Assumed that
apoptosis occurs when the level of p53 is increased
by more than 6 hours (in simulation exceeding the
BIOSTEC2014-DoctoralConsortium
68
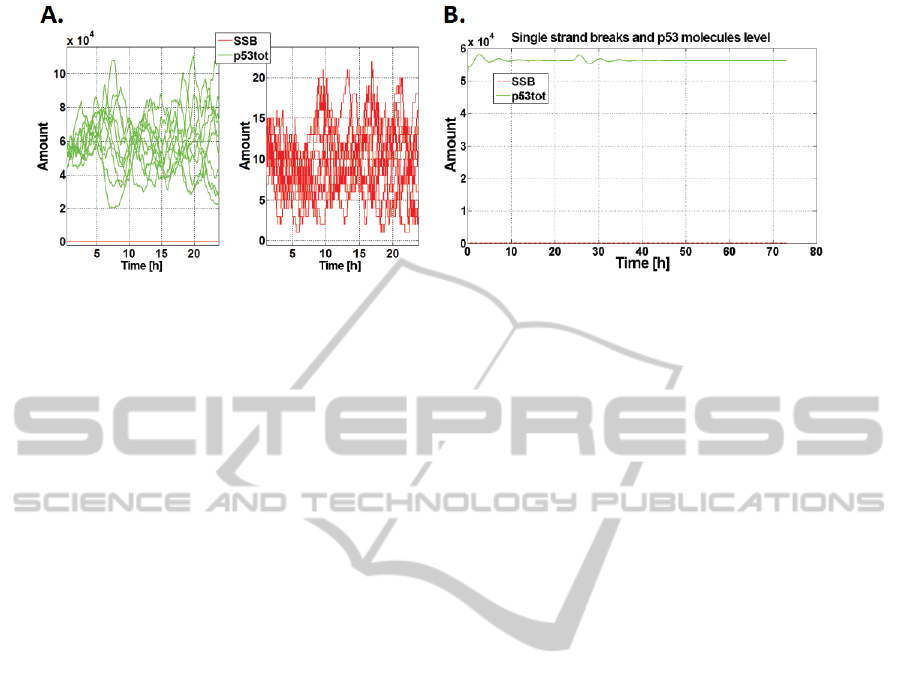
Figure 3: A. Spontaneous DNA damage formation and p53 activation. Result of 10 stochastic simulations; B. Response to
one lesion occurrence. Red - number of SSBs, green - p53 total level.
threshold 2.1·10
5
). Further experiments were run for
the radiation dose of 18 J/m
2
.
4B-D. Disabling Selected Effects
Inhibition of the p53 phosphorylation caused by Chk1
and Chk2 kinases results in the reduction of apoptotic
fraction size (fig. 4B: 17/100 cells for the Chk1 and
14/100 cells to Chk2, for which plot appears almost
identical), and a significant prolongation of DNA re-
pair time. In the case of the inhibition of the ATR-
dependent p53 activation, the apoptotic fraction de-
cline was smaller (size of fraction was 46/100 cells).
A stronger effect was obtained excluding Chk1
(fig. 4C) and Chk2 dependent degradation of Mdm.
None of the cells entered the state of apoptosis. As in
the previous case, less system response change was
observed for the ATR. Apoptotic fraction size was
20/100.
When total Chk1 (fig. 4D) or Chk2 protein kinase
activity is blocked, none of the cells reach the state of
apoptosis. Simulation analysis show the correct func-
tioning of the model stress response which effect is
known from the literature.
1.2.2 Conclusions from Recent Work
ATR module is able to detect a single strand break
caused by UVC appearing in the cell and enhance the
signal so as to cause an increase in p53 protein level.
The threshold of apoptosis in the healthy cell is 18
J/m
2
. However, if the pathway is defective, apoptotic
threshold shifts. Despite extensive damage, the cells
may not die, but transfer incorrect genetic material to
daughter cells (because DNA damage repair takes a
lot longer). This state could potentially be a cause of
cancer and other genetic diseases. Developed model
can be used to study the behavior of cells with spe-
cific mutations without the need for costly and time-
consuming experiments in the laboratory.
1.3 ATM-p53 Model
Independently, the ATM model was created. ATM-
p53 model is based on information from literature re-
garding DSBs detection pathways and role of Wip1
protein (Shimada and Nakanishi, 2013). ATM detec-
tion module regulates p53 phosphorylation via posi-
tive loop. Wip1 creates negative feedbacks for all of
the proteins except Mdm2 in nucleus, where it acti-
vates inactive Mdm2 by dephosphorylation. Activa-
tion of the model takes place by the application of ion-
izing irradiation. The signaling pathway is also stim-
ulated continuously by the small number of damages
that occur spontaneously. Most of the assumptions
about the structure of the model is the same as in ATR
part. ATM model is also an extension of the p53 sig-
naling pathway model (Puszynski et al., 2008). This
model shows that cells with blocked transcription of
Wip1 are more affected to apoptosis. The absence of
PTEN and Chk2 in the model significantly affect the
results of the simulation.
2 OUTLINE OF OBJECTIVES
First of all we planned to combine models ATR-p53
and ATM-p53. This approach is caused by interac-
tions between these paths. However, a more interst-
ing goal is modelling dependence of described above
path on cell cycle phase. We plan to investigate how
the specific cell cycle phase affects the ATM-ATR-
p53 path and the other hand, how the path influences
the cell cycle.
3 RESEARCH PROBLEM
In our study we plan to examine how cell cycle pro-
gression influences DNA damage detection pathway.
DNADamageDetectionanditsImpactontheCellCycle
69

Figure 4: A. Apoptotic death threshold; B-D. Disabling selected effects. Results for 100 stochastic simulations. Solid line -
median; dashed line - upper and lower quartile of results.
The speed of reactions occuring in the cell depends
on the concentration of molecules participating in it.
This concentration differs over time. It is caused e.g.
by the variable volume of the cell depending on its
cell cycle phase. It must be taken into account for
modeling of the ATM-ATR-p53 pathway. Expression
of some proteins and its nucleus or cytoplasm loca-
tion may be different depending on the phase of the
cycle, what will have to be reflected in the proposed
model.
In another look at the model, we plan to examine
how the damage formation caused by stressful factor
in various forms (ionizing radiation, UV light) affects
the cell cycle and for which radiation doses cell cy-
cle will be stopped for DNA damage repair. We plan
to determine which dose will cause apoptosis of cells.
Another important issue is to check how the disabling
of selected interactions in the pathway will affect the
response of the cell.
The purpose of the construction of such a model
is to illustrate the processes occurring in the cell af-
ter a lesion is detected in the different phases of the
cycle, depending on the given force. With this model
we can revise how mutations of the genes encoding
the individual elements of the path and causing inhi-
bition of their activity may influence the behavior of
cells. The model can be used to verify the hypotheses
without the need for costly and long lasting biological
experiments.
4 STATE OF THE ART
4.1 Cell Cycle
The cell cycle consists of two main stages: interphase,
which prepares the cell to the next division and the
division (M phase): mitosis (somatic animal cells) or
meiosis (generative animal cells). Mitosis (which we
will deal more in our model) includes laryokinesis
(division of the cell nucleus) and cytokinesis (divi-
sion of cytoplasm). It results in the separation of the
one cell into two daughter cells (Cooper, 2000). The
major stages of interphase are:
• G
1
Phase - growth phase; biosynthetic processes
in the cell, which were significantly slowed in the
M phase, will be taken up again. In this phase the
synthesis of various enzymes required for DNA
replication in S phase takes place. Length of the
BIOSTEC2014-DoctoralConsortium
70

G
1
phase differs even between cells of the same
species.
• S Phase - begins with DNA synthesis, and lasts
for a similar period of time in all cells. The pur-
pose of the processes taking place in the S phase is
to double the amount of DNA present in the cell.
Each chromosome has been replicated. RNA and
protein synthesis in this phase of the cycle is very
slow (with the exception of histone protein syn-
thesis).
• G
2
phase - in this phase synthesis of proteins is
increased again, mainly those responsible for the
formation of the mitotic spindle (tubulin), which
are necessary for the occurrence of a subsequent
process of mitosis.
• G
0
Phase - eukaryotic cells (especially those fully
differentiated) may move from the G1 phase to the
G0 phase, where they do not undergo divisions
and can remain for a long period of time. The
aging of cells in response to damage is a process
that prevents (without causing apoptosis) transfer
of incorrect genetic material to progeny cells.
4.2 Cell Cycle Checkpoints
Cell cycle checkpoints (fig. 5) are mechanisms which
verify correctness of the DNA. If genetic material is
damaged or not all cellular processes specific for each
phase have been completed, cell cycle progression is
stopped until all will be finished and repaired. If the
damage is too big that could have been repaired, cell
is directed to apoptosis (Cooper, 2000; Shapiro and
Harper, 1999). The main checkpoints in the eukary-
otic cells are:
• G
1
/S checkpoint - at the end of G
1
phase (before
synthesis phase) decision whether cell should di-
vide, delay division or enter a G
0
phase is taken.
• G
2
/M checkpoint - occurs at the end of G
2
phase
(before mitosis) and checks if cell is ready to mi-
tosis (whether mitotic apparatus is fully formed
and whether DNA lesions occur).
4.3 Cyclins, Cyclin-Dependent Kinases,
Cyclin-Dependent Kinase
Phosphatases and
Cyclin-Dependent Kinase Inhibitors
To move to next phases of cell cycle is needed co-
operation of two types of molecules: cyclin and
cyclin-dependent kinases. Cyclins and the cyclin-
dependent kinases (CDKs) form together the active
Figure 5: Cell cycle checkpoints and DNA damage detec-
tion.
heterodimer, where cyclins represent a regulatory unit
and are synthesized in specific phases of the cell cy-
cle in response to various molecular signals. CDKs
play a catalytic function and their expression is inde-
pendent of phase of cell cycle. CDKs upon binding to
cyclins are activated and performs the target protein
phosphorylation reactions, which thus become acti-
vated or inactivated, what coordinate entry into the
next phase of the cell cycle. CDKs are often activated
by cyclin-dependent kinase phosphatases (for exam-
ple Cdc25) - tyrosine phosphatases, which acts by
removing the blocking the CDKs activity phosphate
residues (Orlando et al., 2008). Regulation of CDKs
activity might be performed by CDKs inihibitors (like
p21 encoded by a gene CDKN1) which nhibits the
CDK-cyclin complexes activity. p21 binds to cyclin
E/Cdk2 and cyclin D/Cdk4 complexes and inhibiting
their activity acts as a regulator of the cell cycle in the
G1 phase. p21 gene expression is tightly controlled
by the p53 protein (Gartel and Radhakrishnan, 2005).
4.4 Role of Chk1 and Chk2 in
Checkpoint Mechanism
Between G1 and S phase DNA damage results in the
activation of ATM and ATR and following Chk2 and
Chk1 phosphorylation and next phosphorylation of
p53 and Mdm2, which results in activation and sta-
bilization of p53. Active tetramers act as a transcrip-
tion factor of (among others) p21 protein, which is a
potent inhibitor of cyclin-dependent kinases and pre-
vents cells before the entry to the S phase. In addition,
Chk1 phosphorylates and inactivates Cdc25A phos-
phatase that is essential in CDKs activation.
Blocking of the G2-phase is performed with the
signal transducers ATM or ATR (depending on the
type of damage). They activate Chk1 and Chk2 ki-
nases which phosphorylate Cdc25 phosphatase, caus-
ing its inactivation. This avoids the activation of Cdk1
kinase (encoded by the gene CDC2) necessary to ini-
tiate mitosis (Shapiro and Harper, 1999).
DNADamageDetectionanditsImpactontheCellCycle
71

Figure 6: Schema of new ATM-ATR-p52 and cell cycle pathway.
5 METHODOLOGY
We created scheme od combined ATM-ATR-p53
pathways model with taking into account the key ele-
ments of cell cycle regulation in checkpoints (fig. 6).
Both the scheme and the model are a simplification
of reality what is necessary to enable the modeling of
the pathway.
In signaling pathways modelling we use the basic
laws known from biochemistry: the law of mass ac-
tion and Michaelis-Menten kinetics. The kinetic pa-
rameters for our model we obtain from the results of
biological experiments performed by us and from lit-
erature. Unknown parameters are estimated by fitting
the model to the known data.
As described in caption 1 proposed model will
be based on Haseltine-Rawlings postulate (Haseltine
and Rawlings, 2002) which binds deterministic and
stochastic approach. In our models we use ODE
to simulate fast reactions (in example protein-protein
interactions) and direct Gillespie method (Gillespie,
1977) to simulate slow reactions (enabling genes
and DNA lesions number). In near future we will
have computational system which enables modeling
of variable terms for each of cell cycle phase. We
plan to perform stochastic simulation for population
of cells (in example 1000 cells).
6 EXPECTED OUTCOME
Expected outcome of this research will be to develop
a model illustrating the processes occurring in the cell
associated with the detection of damage and cell cy-
cle progression. The resulting model will allow for
the imaging of various kinds of extortions acting on
BIOSTEC2014-DoctoralConsortium
72

the cells (ioinizing irradiation, UV-light). The result
of model action will be a system response which will
be adequate to the applied force. For healthy cells
at too high dose of damaging agents cells should be
directed to apoptosis, whereas at lower doses cell cy-
cle arrest and repair of damages should occur. For
the population (in example, 1000) modeled stochasti-
cally, desynchronized cells after forcing will show a
division into fractions of cells retained in the appro-
priate phase of the cycle. Constructed model will let
investigate the effect of disable selected interactions
at overall pathway answer. This allows to investigate
how mutation emerged in a cell can influence the cell
cycle and damage detection. Effect will show influ-
ence of abnormalities appearing for example in tumor
cells which are known in the literature.
REFERENCES
Ciccia, A. and Elledge, S. J. (2010). The DNA Damage Re-
sponse: Making It Safe to Play with Knives. Molecu-
lar Cell, 40:179–204.
Cooper, G. M. (2000). The Eukaryotic Cell Cycle. ASM
Press, Washington, D.C, 2 edition.
Gartel, A. L. and Radhakrishnan, S. K. (2005). Lost in Tran-
scription: p21 Repression, Mechanisms, and Conse-
quences. Cancer Research, 65:3980–3985.
Gillespie, D. T. (1977). Exact stochastic simulation of
coupled chemical reactions. The Journal of Physical
Chemistry, 81(25):2340–2361.
Haseltine, E. L. and Rawlings, J. B. (2002). Approxi-
mate simulation of coupled fast and slow reactions
for stochastic chemical kinetics. Journal of Chemical
Physic, 117(15):6959–6969.
Kohn, K. W. (2002). Genomic Instability and DNA Repair.
In Alison, M. R., editor, The Cancer Handbook. 2 edi-
tion.
Orlando, D. A., Lin, C. Y., Bernard, A., Wang, J. Y., So-
colar, J. E. S., Iversen, E. S., J., H. A., and Haase,
S. B. (2008). Global control of cell-cycle transcrip-
tion by coupled CDK and network oscillators. Nature,
453:944–947.
Puszynski, K., Hat, B., and Lipniacki, T. (2008). Oscil-
lations and bistability in the stochastic model of p53
regulation. Journal of Theoretical Biology, 254:452–
465.
Shapiro, G. I. and Harper, J. W. (1999). Anticancer drug
targets: cell cycle and checkpoint control. The Journal
of Clinical Investigation, 104(12):1645–1653.
Shimada, M. and Nakanishi, M. (2013). Response to DNA
damage: why do we need to focus on protein phos-
phatases? Frontiers in Oncology, 3.
DNADamageDetectionanditsImpactontheCellCycle
73
