Forests of Latent Tree Models for Genome-Wide Association Studies
Phan Duc Thanh
LINA, UMR CNRS 6241, Ecole Polytechnique de l’Universit de Nantes, Nantes, France
1 CONTEXT
With the finalization of the Human Genome Project in
2003, it was confirmed that any two individuals share,
on average, 99.9% of their genome with each other.
It is the sole 0.1% genetic variations that explain why
individuals are physically different or inherit a greater
risk of contracting disorders, such as heart disease or
cancer. Therefore, identifying the genetic factors un-
derlying disease can potentially play a crucial role in
developing new treatments and has been one of the
main focus of human genetics research during the last
thirty years (Hechter, 2011). Among different ap-
proaches that have been proposed, association study
stands out as one of the most successful path, even
though its potential is yet to be fully tapped.
During the past decades, a great deal of effort has
been put into the investigation of heritable suscep-
tibility to complex diseases which, contrary to rare
monogenic disorders, are thought to be affected by,
not a single one, but multiple genetic variants. In ef-
fect, according to a hypothesis known as common dis-
eases - common variants (CDCV), it is conjectured
that most of the risk of common disorders, such as
cancers, could be explained by common variations in
several genes. Since these variants are common, they
are susceptible to detection using association studies.
Traditionally, the strategies for association study
involve performing analysis with only a small num-
ber of loci (DNA locations) pre-chosen with the help
of prior knowledge. For example, fine-mapping stud-
ies are conducted only in a pre-selected candidate
region of 1-10Mb (one Mb equals 10
6
nucleotides).
Thanks to new advances in techniques for genotyping
and sequencing genomes, researchers started to work
on seeking genetic variations potentially associated
to common diseases throughout the entire genome
(GWAS). In the following years, the HapMap Project
and its successor, the 1000 Genomes Project, were
launched with the hope to establish a catalogue of
human genome regions in which people of different
populations have differences.
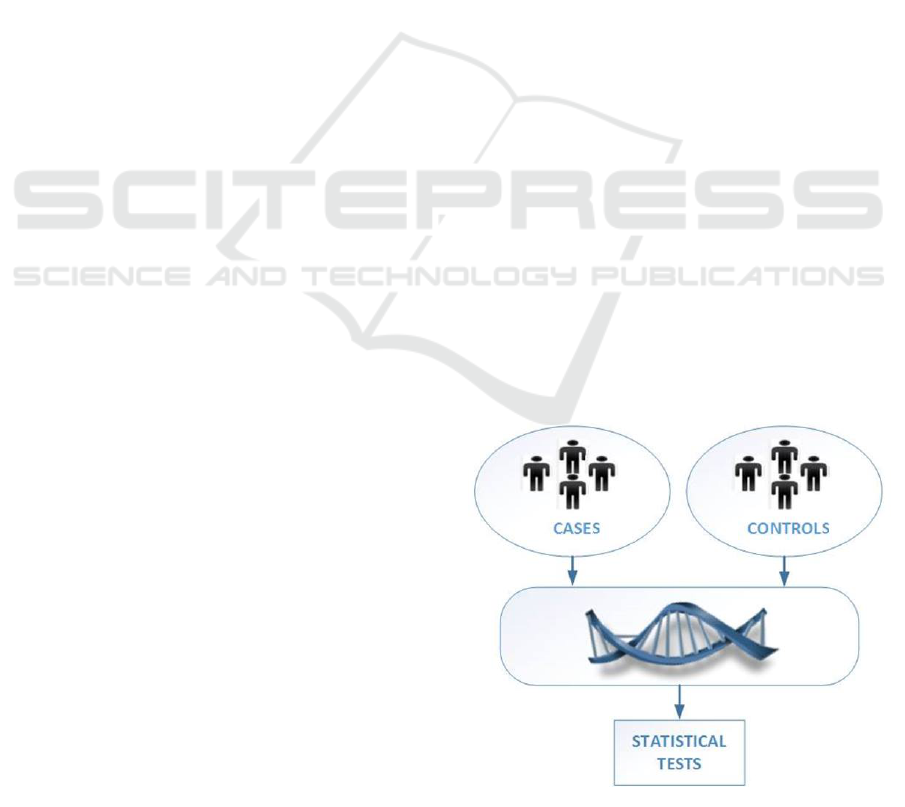
In a nutshell, GWASs seek to identify combi-
nations of markers (DNA sequence with a known
location) whose frequency vary systematically be-
tween individuals with different disease states (Bald-
ing, 2006). Its most basic form consists in comparing
allele frequencies in cases and controls, as depicted
in Figure 1, on data of generally large size (hundreds
of millions genetic variants from thousands of peo-
ple). The goal is to identify the loci on the genome
for which the distributions of observations are signif-
icantly different, using statistical tests (e.g. the Chi
2
test). In that case, we have reasons to believe that
the gene affecting the trait might be located some-
where in the neighborhood of the pinpointed region.
The unit variants, called single-nucleotide polymor-
phisms (SNPs) (single base-pair changes in the DNA
sequence), are very often used as markers in GWASs.
SNPs are by far the most abundant type of variant in
human.
Figure 1: GWAS procedure.
Apart from identifying disease-related factors, an-
other motivation for GWASs is the increasing deep
societal mutation observed in Western countries. In
France, about 12% of the gross domestic product are
currently dedicated to the public health expenditure,
whereas this percentage was only 3% 30 years ago.
For a main part, this ever increasing share of pub-
lic health expenditure is to be related to the gain in
74
Duc Thanh P..
Forests of Latent Tree Models for Genome-Wide Association Studies.
Copyright
c
2014 SCITEPRESS (Science and Technology Publications, Lda.)

longevity, which favours the emergence of chronic
diseases by elderly subjects. Therefore, a better un-
derstanding of gene susceptibility to pathologies is
expected to better control public health expenditure.
Other applications of GWASs can be found in
pharmacology where scientists seek to correlate pat-
terns of variations associated with phenotypes such as
adverse drug responses and drug metabolism or par-
ticularly in personalized medicine where healthcare
decisions can be guided by individuals’ genetic pro-
file.
Many evidences obtained have led to believe that
the CD-CV hypothesis holds. It is then essential to
obtain the most common patterns of human genetic
variation, hence the motivation for the Human Hap-
lotype Map (HapMap) project and its successor, the
1000 Genomes Project (a haplotype can be informally
described as a cluster of nearby SNPs). The main goal
of these international efforts is to identify and cata-
logue variations and their relations across the entire
human genome from various populations. From these
databases, we are able to study a known phenomenon
called linkage disequilibrium (LD), referring to the
dependence between SNPs at two or more sites.
LD plays a role of huge importance in GWASs for
various reasons. A well-designed study will have a
good chance of including one or more SNPs that are in
strong LD with a common causal variant. LD reflects
the blurring of the ancestral genome, mainly but not
only due to combinations and mutations. Standard
statistical approaches that do not take into account this
type of correlation between variables will not work
well on genome-wide data.
The explosion of complex GWAS data has re-
quired the development of new analysis methods for
dealing with challenges regarding, among others, sta-
tistical power increase and false discovery rate con-
trol. Within bioinformatics in particular, probabilistic
graphical models (PGMs) are considered as powerful
machine learning approaches thanks to their capacity
for capturing complex relationships and dealing with
high-dimensionality. In this regard, the main purpose
of this thesis is to design PGM-based GWAS strate-
gies that are capable of effectively finding suscepti-
bility to complex diseases.
The rest of this paper is organized as follows. We
will first provide a brief overview of various LD mod-
elling approaches relying on machine learning tech-
niques, and among them on PGM-based methods; we
will then discuss different challenges that may arise;
the outline of desired objectives we aim to achieve, as
well as the research methodology, will also be given,
followed by a presentation of the current research
stage.
2 STATE OF THE ART
The complex structure of LD in the human genome
was revealed by the HapMap project. More specif-
ically, it is claimed that LD is highly structured
into the so-called “haplotype block structure” regions
(Patil et al., 2001) where statistical dependences be-
tween contiguous markers (called blocks) alternate
with shorter regions characterized by low statistical
dependences. In addition to complexity, a systemic
whole-genome analysis with high-density data typ-
ically involves a large number of variables, which
poses a tremendous challenge in terms of scalability.
Interestingly, LD may offer a solution to dimen-
sionality reduction. Relying on the “haplotype block
structure”, various approaches have been proposed to
perform multi-SNP association test with haplotypes,
i.e. inferred data underlying genotypic data such as
in (Schaid, 2004), or partitioning the genome accord-
ing to spatial correlation (Pattaro et al., 2008). By
contrast, the method proposed in (Han et al., 2008)
chooses to select SNPs informative about their con-
text - or SNP tags. The HaploBuild software pro-
gram (Laramie et al., 2007) allows the construction
of more biologically relevant haplotypes that are not
constrained by arbitrary length, thus making it able to
learn ”haplotype cluster structure”.
In general, LD exhibited among physically close
loci is stronger than LD between SNPs that are farther
apart. In other words, LD decays with distance. Lim-
itations of previous stated methods include not taking
into account long-range dependences. Besides, these
methods do not consider the fuzzy nature of LD which
refers to the fact that LD block boundaries are not ac-
curately defined over the genome.
Probabilistic graphical models (PGMs), due to
their appealing characteristics, represent an appropri-
ate framework and have gained indeed significant at-
tention from researchers to analyse LD .
2.1 Probabilistic Graphical Models in
Linkage Disequilibrium Modelling
Graphical models provide rich families of graph-
based probabilistic models of joint multivariate prob-
ability distributions that capture properties of con-
ditional independence between variables (Friedman
et al., 2000). In a graph, nodes represent the variables
and edges denote direct dependences between these
variables. There are two main classes of probabilistic
graphical models, namely Bayesian Networks (BNs)
and Markov Random Fields (MRFs).
As pointed out, PGMs allow to capture complex
dependences between SNPs (due to LD). In addition,
ForestsofLatentTreeModelsforGenome-WideAssociationStudies
75

we can also integrate useful prior domain knowledge
to enhance the model quality as well as to improve
the performance of its construction. For example, as
PGMs can describe very well local dependences, by
limiting the network of dependences for each node
within a certain physical range, we can dramatically
reduce the model’s complexity (Verzilli et al., 2006).
However, although PGMs had long been widely rec-
ognized as a powerful formalism in the bioinformatics
domain in gene expression studies and linkage analy-
sis, they received much less attention in genetic asso-
ciation studies up to recently.
2.1.1 Markov Random Fields
MRFs are a class of PGMs where undirected graphs
are used to represent dependences. The study of ap-
plying MRFs in modelling LD has been rather active
since first paper by Alun Thomas et al. (Thomas and
Camp, 2004) in which they restricted the computa-
tion of the joint distribution to a tractable subclass
of MRFs called decomposable (or equivalently trian-
gulated). The idea is to use a simulated annealing
search strategy which, given a decomposable graph,
computes efficiently the score of one of its neighbour
graphs.
Also relying on Decomposable MRFs (DMRF),
Verzilli et al. in (Verzilli et al., 2006) proposed to
model haplotype blocks explicitly as cliques. More-
over, each clique is labelled with a boolean value
(1/0) indicating the existence of at least one edge be-
tween some vertex in the clique and the phenotype
(i.e. disease) vertex. Since checking the decompos-
ability property is computationally demanding, the
authors opted to select appropriate moves to preserve
this property while browsing the search space of DM-
RFs.
2.1.2 Bayesian Networks
Bayesian Networks, on the other hand, employ
acyclic directed graphs to specify joint distribution
over the variables. The models within this class can
be further categorized into two different paradigms:
with and without latent variables (LV). An LV repre-
sents factors that exist but are unobservable and may
induce correlations between observed variables that
do not correspond to direct causal relations (Kollar
and Friedman, 2009).
Belonging to the former category, BNTagger is
a method proposed by Lee and Shatkay in (Lee and
Shatkay, 2006) for SNP tag selection. To efficiently
learn the structure, the authors opted for a greedy
search strategy with random restarts. The final aim
of this modelling is to determine a subset of indepen-
dent and highly predictive SNPs.
BN models with LVs have also been studied to
model and exploit LD. The general principle com-
prises using the structural expectation maximization
(SEM) algorithm (Friedman et al., 2000) to infer the
structure as well as the model parameters. Nefian, for
instance, proposed a method (Nefian, 2006) based on
the concept of Embedded Bayesian Networks (EBN).
EBNs are hierarchical BNs where a subset of ob-
served nodes have as parent only one latent node, to
form a latent class model (LCM).
The two-layer model employed by Nefian can
possibly be enriched with SNP dependences. Ne-
fian’s block-based approach involves first splitting the
genome into contiguous windows (i.e. blocks), then
learning an LCM for each window. Afterwards, SEM
is applied to learn dependences between SNPs and be-
tween LVs. However, this model imposes that vari-
ables in the same LCM must be spatially contiguous
on the genome. Further, the window size is fixed to a
small value. A consequence is a severe lack of flexi-
bility. To address this shortcoming, another two-layer
BN was proposed by Zhang and Ji (Zhang and Ji,
2009) which can be described as a mere set of non-
connected LCMs. This model relaxes several condi-
tions of the previous one, including that the SNPs in
the same cluster are not required to be spatially con-
tiguous and that the cluster sizes can vary. However,
the number of clusters is a parameter which has to be
specified.
2.2 Scalable Methods
None of these aforementioned methods scales well
with the number of SNPs and number of individu-
als in the observed population. Several methods have
been proposed to address this problem in the GWAS
context. Among those, Hidden Markov models
(HMMs) were used by Scheet and Stephens (Scheet
and Stephens, 2006) to infer haplotypes. Therein,
the latent states correspond to ancestral haplotypes.
This model can deal with either block-like LD struc-
ture or gradual decline of LD with distance. It is
implemented in the well-known accurate and scal-
able fastPHASE program (one thousand individuals
and hundreds of thousands of genetic markers). Be-
sides, in contrast with block-based models, this model
provides flexibility since it allows ancestral haplotype
membership to change at any SNP site.
In contrast with Scheet and Stephen, Browning
and Browning proposed the variable-length Markov
chain (VLMC) where no pre-specification of the
structure, such as the number of ancestral haplotypes,
BIOSTEC2014-DoctoralConsortium
76

is required (Browning and Browning, 2009). VLMCs
allow the memory length to vary along the chain. This
work resulted in the widely used BEAGLE tool which
scales particularly well with datasets of hundreds of
thousands of markers describing thousands of individ-
uals.
Another possibility to obtain scalability is through
interval graphs in which each vertex may be associ-
ated with an interval of the genome and edges connect
any two strictly overlapping intervals. These graphs
can be proved to be decomposable, thus resolving
the non-decomposability issue (Thomas, 2009). On
the other hand, local computation of the likelihood is
implemented during the Markov chain Monte Carlo
(MCMC) sampling devoted to interval updating. In-
tervals are particularly appealing to account for LD
extent around a locus. Finally, more recent works ex-
ploiting graph theory properties were shown to actu-
ally help achieve notable performance gain in DMRF
structure learning (Abel and Thomas, 2011; Thomas
and Green, 2009).
Unfortunately, none of the aforementioned ap-
proaches lends itself to incorporate data to reinforce
evidence for genotype-disease association identifica-
tion, in a GWAS context. In addition, the model
choice for VLMC in BEAGLE cannot take into con-
sideration long-range or hierarchical dependences in
genotype data.
In conclusion, even though PGMs have been used
and have shown promises as an alternative to tradi-
tional GWAS analysis methods, they still have various
limitations to address. In addition, only a few number
of model based strategies have been proposed up to
now to perform GWASs.
3 RESEARCH PROBLEMS
Despite the early successes of GWASs, various chal-
lenges remain to be addressed. First of all, the
task of analyzing SNP combinations across the en-
tire genome is very challenging computationally, due
to the large number of SNPs in high-density GWAS
data. Further, the complexity increases exponentially
with the number of SNPs to consider (this issue is
known as “the curse of dimensionality”), which has
necessitated the development of innovative methods.
Besides, as the number of SNPs is usually signifi-
cantly bigger than the number of individuals, we often
face the so-called small n-large p problem, for which
traditional statistical methods cannot be applied. In
addition to the enormous quantity of variables, the re-
lationships among variables (i.e. SNPs) as well as be-
tween disease susceptibility genotypes and diseases
are very complex and far from being fully understood.
Failing to take into consideration these types of high-
order, non-linear correlations, a GWAS strategy will
likely be unable to efficiently identify the causal vari-
ants.
On the other hand, a good study design should
take account of additional prior knowledge, for var-
ious reasons. In effect, even if the computational
methods can identify correctly disease-related SNPs,
it can be translated into improving health care deci-
sion know-how only if the context of biology is taken
into account (Moore et al., 2010). These kinds of do-
main knowledge can come from various forms such
as prior biological findings or supplementary data
sources. For instance, gene ontology, gene-gene in-
teractions and transcription factors’ targets data can
potentially provide useful information.
Furthermore, the evaluation of the innovative
GWAS strategies will require the fast generation of
realistic genome-wide simulated data. For this pur-
pose, a gold standard is HapGen (Su et al., 2011)
which consists in simulating genotypes given the phe-
notypes. However, because it generates both geno-
typic and phenotypic data for each simulation, it is
considered not fast enough for our needs.
Last but not least, conducting effectively exper-
iments on massive genome-wide datasets demands
high performance software codes. This is a com-
mon issue for many fields which require dealing with
large-scale computation. The implementation should
be resource-optimized while able to maintain a certain
degree of user-friendliness to facilitate the communi-
cation of results. Moreover, it should be noted that it
is not always easy to provide output that is simulta-
neously visual and easy to navigate. This last point
is relevant due to the needs for communicating results
with other collaborators of different backgrounds (e.g.
geneticists, physicians).
4 OUTLINE OF OBJECTIVES
Since the first successful GWAS in 2005 (Klein et al.,
2005), the amount of yearly published studies has
constantly increased, reaching more than 2300 stud-
ies in 2011. Nowadays, GWAS has evolved to be a
major method to identify genetic risk factors for dis-
eases. As pointed out, PGMs have proven to be very
promising for the statistical analysis of GWAS data
but their potential is yet to be fully achieved. In this
thesis work, we wish to explore this path, develop-
ing novel computational PGM-based GWAS strate-
gies for unravelling the underlying genetic structure
of common disorders.
ForestsofLatentTreeModelsforGenome-WideAssociationStudies
77

Carried out at the KOD team of the LINA labora-
tory and in the framework of the ANR project named
SAMOGWAS, the objectives of this thesis are numer-
ous and varied, which include but are not limited to:
• model the genetic data at the genome scale and de-
sign the corresponding method to construct such
models
• develop, evaluate and compare several PGM-
based GWAS strategies,
• apply the best strategy on real biological data.
The first goal is to establish a framework for
modelling genotype data, based on a class of PGMs
called forests of Latent Tree Models. Within this
framework, we will then design and compare several
GWAS strategies. Central to our design is the incor-
poration of domain knowledge to enhance the GWAS
strategies. Besides, we aim to address the lack of
methods for GWAS strategy evaluation and valida-
tion. Finally, in the context of GWASs, the computa-
tion involved typically takes a long time, ranging from
days to weeks and even months (Fabregat-Traver and
Bientinesi, 2012). As a consequence, we also plan to
deliver highly-optimized applications to realize effec-
tively the work.
5 METHODOLOGY
This multidisciplinary and interdisciplinary work will
be carried out in an incremental manner, in close col-
laboration with multiple partners (geneticists, genetic
epidemiologists, computer scientists and mathemati-
cians). The main idea is to establish a modelling
framework based on the Forests of Latent Tree Mod-
els (F models).
First introduced by Zhang and initially called hi-
erarchical latent class models (Zhang, 2004), F mod-
els are tree-structured Bayesian networks where leaf
nodes are observed while internal nodes are not. In
latent tree models (LTMs), multiple latent variables
organized in a hierarchical way allow to depict a large
variety of relations encompassing local to higher-
order dependences, as depicted in figure 2. The sur-
vey in (Mourad et al., 2013) provides an exhaustive
review of the different classes of methods used for
learning the structure of LTMs. Among these meth-
ods, there is one simple procedure which relies on ag-
glomerative hierarchical clustering and consists in it-
erating two main steps: (i) discovering cliques of de-
pendent variables; (ii) synthesizing cliques’ informa-
tion by latent variables (Mourad et al., 2010).
A first version of this specific learning algo-
rithm has been shown to be tractable on bench-
marks describing 100000 variables for 2000 individu-
als (Mourad et al., 2011). The theoretical complexity
was proved to scale linearly with the number of SNPs
and quadratically with the number of individuals. The
versatile LTMs offer an adapted framework to encode
the fuzzy nature of linkage disequilibrium blocks. Be-
sides, as previously shown, by introducing latent vari-
ables, LTMs can enable data dimensionality reduc-
tion. The scalability comes from the reduction of the
data dimension, due to the subsumption of variables
through latent variables. The flexibility comes from
the generality of the forest (i.e. it is not constrained to
be a binary topology; moreover, the (discrete) latent
variables may have different cardinalities).
Figure 2: Latent tree model. The light shade indicates the
observed variables whereas the dark shade points out the
latent variables.
In addition to PGMs, Random Forest (RF) is an-
other Machine Learning technique which is often em-
ployed in GWASs, thanks to its advantage in analyz-
ing high dimensional data. A specific adaptation of
the Random Forest method has been investigated by
the GIGA-R partner of the SAMOGWAS project, to
perform GWASs, leading to the T-tree model (Botta
et al., 2008). In contrast with the standard applica-
tion to GWASs of a Random Forest-based method, the
idea is to exploit the haplotype block structure by re-
placing SNP-based splits with haplotype block-based
splits in this decision tree-based method. In addition
to genotyped cases and controls, this algorithm thus
needs as input a decomposition of the set of SNPs into
haplotype blocks.
We also observed that in the F model, each la-
tent variable represents a haplotype block while a de-
composition of the set of SNPs into haplotype blocks
is requested to run the T-tree-based approach. Con-
versely, the haplotype block importance computed by
the T-tree-based method could be exploited by the F
model to target a potentially causal block for the stud-
ied disease. Thus, a combination of these two models,
where the clusters identified by F models will serve as
prior knowledge for T-tree models, looks like a very
promising perspective.
BIOSTEC2014-DoctoralConsortium
78

On top of this, various GWAS strategies will be
designed based on the aforementioned models. For
example, a GWAS strategy based on F-models should
involve traversing the forest for finding the most sig-
nificantly associate nodes. A solution for this consists
in running a best-first search traversal for any sub-
tree rooted in a latent variable significantly associated
with the disease.
In parallel, various sets of real-life data will be
collected. These datasets will be used for the final
validation of the results which will then be interpreted
together with biologists. We will also incorporate
to our GWAS strategies supplementary transcriptome
data and additional knowledge from gene ontologies
and from gene annotation databases, with the help of
our collaborators. This incorporation will likely al-
low the enrichment of the models as well as the cross-
confirmation of putative associations between genetic
factors and disease.
Regarding the evaluation of model-based strate-
gies, we need a method that can quickly and re-
liably generate consistent genotypic, transcriptomic
and phenotypic data. First, the current standard meth-
ods for GWAS data simulation are not very efficient
because they generate both genotypic and phenotypic
data for each simulation. In contrast, when a GWAS
relies on the time-consuming construction of a com-
plex model of the LD (i.e. the F model), we wish
to generate only simulated phenotypes while keeping
the same genotypic data for all simulations. For this
purpose, the key is to sequentially sample the phe-
notypes according to a probability distribution that
accounts for the total number of cases to be gener-
ated and the current number of cases already gener-
ated. Second, generating both GWAS data and sim-
ulated transcriptomic data in a consistent way repre-
sents a real challenge. For this purpose, we will inves-
tigate several adaptations of the previously described
method.
Last but not least, as previously stated, our
work’s realization needs to be robust and efficient
for handling computation-intensive tasks. The idea
to achieve performance-optimized implementation re-
lies mainly on CPU-based parallelization of code.
The applications will be able to be deployed on multi-
core as well as grid computing systems. Finally, since
F Models are based on graphs, several approaches
for effectively visualizing and manipulating F mod-
els will be explored. One of them relies on the Tulip
visualization tool designed to display and annotate
large graphs (Auber, 2004). Such visual representa-
tions may enable better analysis of results by the end
users.
6 STAGE OF THE RESEARCH
Started in October 2013, this thesis will be carried on
for the next 3 years within the KOD team at the Ecole
Polytechnique de l’Universit
´
e de Nantes, under the
supervision of Christine Sinoquet and Philippe Leray.
Currently at the early stage, we are working within the
methodology previously stated on several aspects in-
cluding the implementation of the framework. We are
now designing and implementing a framework opti-
mized for high-performance computing, a crucial ne-
cessity for deep GWAS analysis. The idea consists
mainly in using parallel C++ coding for CPU-based
systems, facilated by OpenMP (Dagum and Menon,
1998) and Open MPI (Gabriel et al., 2004). OpenMP
is an application programming interface (API) that
supports multi-platform shared memory multipro-
cessing whereas Open MPI is a popular implemen-
tation of the standardized Message Passing Interface
(MPI) allowing to run applications across computer
clusters. We also rely on a Bayesian Network library
called ProBT, provided by the ProbaYes partner in the
SAMOGWAS project (www.probayes.com). Once
finished, we will use this application as back-end for
carrying out experiments to evaluate F-Model-based
GWAS on simulated data (spring 2014). Front-end
applications for visualizing and manipulating analysis
results are also going to be provided. For the former
task, the genome-wide visualization of the F models
can be implemented in C++ with the help of the Tulip
library (http://tulip.labri.fr/TulipDrupal/). In parallel
with these implementation and test tasks, in the first
half of 2014, a thorough methodological work focus-
ing on additional data integration will be conducted
in collaboration with the INSERM partner (i.e. biolo-
gists).
7 EXPECTED OUTCOME
Already considered as an important bio-medical re-
search interest, GWASs are still a relatively young
area which have generated numerous and varied dif-
ficult challenges. After this thesis work, we hope
to obtain a proven GWAS model-based integrative
strategy and a novel simulation strategy satisfying
the conditions previously mentioned. We also antici-
pate to have a reliable high-performance software im-
plementation of both the model learning algorithms
and the model-based GWAS strategies. The back-
end applications will be deployed on the GIGA-R and
INSERM grid computing platforms to provide high
quality analysis services. Above all, this multidisci-
plinary work is expected to help expand our knowl-
ForestsofLatentTreeModelsforGenome-WideAssociationStudies
79

edge about genetic variants that influence the suscep-
tibility to complex diseases. In the process, it may
help gain progress across multiple-domains, includ-
ing:
• machine learning and data mining: statistical
models and techniques for dealing with GWAS
data that take account high-dimensionality and
variable correlation will be designed; methods for
the improvement of models through integrating
additional knowledge will be proposed;
• medicine: the methods designed intend to bring
new evidence of genetic disease susceptibility
as well as individual genetic susceptibility to
drugs, thus offering perspectives for personalized
medicine;
• public health: our work will contribute to account-
ing for the societal evolution trend toward early
gene susceptibility detection to improve preven-
tion or surveillance;
• economy: The animal and plant biology domains
are also concerned with respect to the selection of
phenotypes of interest in agronomy as well;
• high-performance computing for GWASs: cutting
edge techniques for implementing GWAS strate-
gies and large-scale machine learning approaches
will be implemented.
REFERENCES
Abel, H. J. and Thomas, A. (2011). Accuracy and computa-
tional efficiency of a graphical modeling approach to
linkage disequilibrium estimation. Statistical applica-
tions in genetics and molecular biology, 10(1).
Auber, D. (2004). Tulip: A huge graph visualization frame-
work. In Graph Drawing Software, pages 105–126.
Springer.
Balding, D. J. (2006). A tutorial on statistical methods for
population association studies. Nature Reviews Ge-
netics, 7(10):781–791.
Botta, V., Hansoul, S., Geurts, P., and Wehenkel, L. (2008).
Raw genotypes vs haplotype blocks for genome wide
association studies by random forests. In Proc. of
MLSB 2008, second workshop on Machine Learning
in Systems Biology.
Browning, B. L. and Browning, S. R. (2009). A unified ap-
proach to genotype imputation and haplotype-phase
inference for large data sets of trios and unrelated in-
dividuals. The American Journal of Human Genetics,
84(2):210–223.
Dagum, L. and Menon, R. (1998). Openmp: an industry
standard api for shared-memory programming. Com-
putational Science & Engineering, IEEE, 5(1):46–55.
Fabregat-Traver, D. and Bientinesi, P. (2012). Comput-
ing petaflops over terabytes of data: The case of
genome-wide association studies. arXiv preprint
arXiv:1210.7683.
Friedman, N., Linial, M., Nachman, I., and Pe’er, D. (2000).
Using bayesian networks to analyze expression data.
Journal of computational biology, 7(3-4):601–620.
Gabriel, E., Fagg, G. E., Bosilca, G., Angskun, T., Don-
garra, J. J., Squyres, J. M., Sahay, V., Kambadur, P.,
Barrett, B., Lumsdaine, A., et al. (2004). Open mpi:
Goals, concept, and design of a next generation mpi
implementation. In Recent Advances in Parallel Vir-
tual Machine and Message Passing Interface, pages
97–104. Springer.
Han, B., Kang, H., Seo, M. S., Zaitlen, N., and Eskin, E.
(2008). Efficient association study design via power-
optimized tag snp selection. Annals of human genet-
ics, 72(6):834–847.
Hechter, E. (2011). On genetic variants underlying common
disease. PhD thesis, Oxford University.
Klein, R. J., Zeiss, C., Chew, E. Y., Tsai, J.-Y., Sackler,
R. S., Haynes, C., Henning, A. K., SanGiovanni, J. P.,
Mane, S. M., Mayne, S. T., et al. (2005). Complement
factor h polymorphism in age-related macular degen-
eration. Science, 308(5720):385–389.
Kollar, D. and Friedman, N. (2009). Probabilistic graphical
models: principles and techniques. The MIT Press.
Laramie, J. M., Wilk, J. B., DeStefano, A. L., and Myers,
R. H. (2007). Haplobuild: an algorithm to construct
non-contiguous associated haplotypes in family based
genetic studies. Bioinformatics, 23(16):2190–2192.
Lee, P. H. and Shatkay, H. (2006). Bntagger: improved tag-
ging snp selection using bayesian networks. In ISMB
(Supplement of Bioinformatics), pages 211–219.
Moore, J. H., Asselbergs, F. W., and Williams, S. M. (2010).
Bioinformatics challenges for genome-wide associa-
tion studies. Bioinformatics, 26(4):445–455.
Mourad, R., Sinoquet, C., and Leray, P. (2010). Learning
hierarchical bayesian networks for genome-wide asso-
ciation studies. In Proceedings of COMPSTAT’2010,
pages 549–556. Springer.
Mourad, R., Sinoquet, C., and Leray, P. (2011). A hier-
archical bayesian network approach for linkage dis-
equilibrium modeling and data-dimensionality reduc-
tion prior to genome-wide association studies. BMC
bioinformatics, 12(1):16.
Mourad, R., Sinoquet, C., Zhang, N. L., Liu, T., Leray, P.,
et al. (2013). A survey on latent tree models and ap-
plications. J. Artif. Intell. Res.(JAIR), 47:157–203.
Nefian, A. V. (2006). Learning snp dependencies using em-
bedded bayesian networks. In IEEE Computational
Systems, Bioinformatics Conference, pages 1–6.
Patil, N., Berno, A. J., Hinds, D. A., Barrett, W. A.,
Doshi, J. M., Hacker, C. R., Kautzer, C. R., Lee,
D. H., Marjoribanks, C., McDonough, D. P., et al.
(2001). Blocks of limited haplotype diversity revealed
by high-resolution scanning of human chromosome
21. Science, 294(5547):1719–1723.
Pattaro, C., Ruczinski, I., Fallin, D., and Parmigiani, G.
(2008). Haplotype block partitioning as a tool for
dimensionality reduction in snp association studies.
BMC genomics, 9(1):405.
BIOSTEC2014-DoctoralConsortium
80

Schaid, D. J. (2004). Evaluating associations of haplotypes
with traits. Genetic epidemiology, 27(4):348–364.
Scheet, P. and Stephens, M. (2006). A fast and flexible
statistical model for large-scale population genotype
data: applications to inferring missing genotypes and
haplotypic phase. The American Journal of Human
Genetics, 78(4):629–644.
Su, Z., Marchini, J., and Donnelly, P. (2011). Hapgen2:
simulation of multiple disease snps. Bioinformatics,
27(16):2304–2305.
Thomas, A. (2009). Estimation of graphical models whose
conditional independence graphs are interval graphs
and its application to modelling linkage disequilib-
rium. Computational statistics & data analysis,
53(5):1818–1828.
Thomas, A. and Camp, N. J. (2004). Graphical modeling of
the joint distribution of alleles at associated loci. The
American Journal of Human Genetics, 74(6):1088–
1101.
Thomas, A. and Green, P. J. (2009). Enumerating the junc-
tion trees of a decomposable graph. Journal of Com-
putational and Graphical Statistics, 18(4):930–940.
Verzilli, C. J., Stallard, N., and Whittaker, J. C. (2006).
Bayesian graphical models for genomewide associa-
tion studies. The american journal of human genetics,
79(1):100–112.
Zhang, N. L. (2004). Hierarchical latent class models for
cluster analysis. The Journal of Machine Learning
Research, 5:697–723.
Zhang, Y. and Ji, L. (2009). Clustering of snps by a
structural em algorithm. In Bioinformatics, Systems
Biology and Intelligent Computing, 2009. IJCBS’09,
pages 147–150. IEEE.
ForestsofLatentTreeModelsforGenome-WideAssociationStudies
81
