
Discovering New Proteins in Plant Mitochondria by
RNA Editing Simulation
Fabio Fassetti
1
, Claudia Giallombardo
2
, Ofelia Leone
1
, Luigi Palopoli
1
, Simona E. Rombo
2,∗
and Adolfo Saiardi
3
1
DIMES - Universit
`
a della Calabria, Rende (CS), Italy
2
Dipartimento di Matematica e Informatica, Universit
`
a degli Studi di Palermo, Palermo, Italy
3
LMCB, MRC, Cell Biology Unit & Department of Developmental Biology, University College, London, U.K.
Keywords:
Sequence Analysis, Editing Simulation, ORF Sequences, Plant mtDNA, Protein Prediction.
Abstract:
In plant mitochondria an essential mechanism for gene expression is RNA editing, often influencing the syn-
thesis of functional proteins. RNA editing alters the linearity of genetic information transfer. Indeed it causes
differences between RNAs and their coding DNA sequences that hinder both experimental and computational
research of genes. Therefore common software tools for gene search, successfully applied to find canonical
genes, often fail in discovering genes encrypted in the genome of plants.
Here we propose a novel strategy useful to identify candidate coding sequences resulting from possible editing
substitutions. In particular, we consider c → u substitutions leading to the creation of new start and stop codons
in the mitochondrial DNA of a given input organism. We try to mimic the natural RNA editing mechanism,
in order to generate candidate Open Reading Frame sequences that could code for novel, uncharacterized
proteins. Results obtained analyzing the mtDNA of Oryza sativa are supportive of this approach, since we
identified thirteen Open Reading Frame sequences transcribed in Oryza, that do not correspond to already
known proteins. Five of the corresponding amino acid sequences present high homologies with proteins al-
ready discovered in other organisms, whereas, for the remaining ones, no such homology was detected.
1 INTRODUCTION
In mitochondria and chloroplasts of flowering plants,
the linearity of genetic information is interrupted by
mechanisms that increase protein variability. Such
mechanisms can alter the RNA transcript so that their
final primary nucleotide sequence results quite dif-
ferent from the corresponding DNA sequence. The
most common among these mechanisms is post-
transcriptional mRNA editing, consisting in enzy-
matic modification of nitrogenous bases, almost ex-
clusively Cytidine to Uridine transformation (Take-
naka et al., 2008). Most RNA editing events are found
in the coding regions of mRNAs and usually at first
and second position of codon, so that the deriving
amino acid is often different from that specified by
the corresponding unedited codon (Gray et al., 1992).
Editing can also create new start and stop codons
(Hoch et al., 1991), (Wintz and Hanson, 1991) and it
can occur in introns (Brennicke et al., 1999) and other
*
Corresponding author
non translated regions (Schuster et al., 1990). The
use of editing to generate aug start codons might rep-
resent another level of regulatory control of gene ex-
pression: introducing a translational start codon could
make an mRNA accessible for protein synthesis (Tak-
enaka et al., 2008).
Specifically, in plant mitochondria, RNA editing
is essential for gene expression. In many cases this
mechanism completes the genomic information and
is essential to the creation of a functional open read-
ing frame (Regina et al., 2002). Given the physiologi-
cal importance of RNA, identification of sites of RNA
editing is essential for molecular, biochemical and
phylogenetic studies in plant mitochondria. Exper-
imental analysis, made comparing RNA transcripts
and genomic DNA sequences, is the more exhaus-
tive way, but it is also expensive and time consuming.
A collection of all sequences post-transcriptionally
modified by RNA editing from many organisms, re-
covered from primary databases and literature, is
available on the RNA editing database REDI (Pi-
cardi et al., 2007). Computational approaches have
182
Fassetti F., Giallombardo C., Leone O., Palopoli L., Rombo S. and Saiardi A.
Discovering New Proteins in Plant Mitochondria by RNA Editing Simulation.
DOI: 10.5220/0005664901820189
In Proceedings of the 9th Inter national Joint Conference on Biomedical Engineering Systems and Technologies (BIOINFORMATICS 2016), pages 182-189
ISBN: 978-989-758-170-0
Copyright
c
2016 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

been used to predict sites of RNA editing, based ei-
ther on statistical methods (Bundschuh, 2004) or on
evolutionary considerations. The latter ones are based
on the observation that often the final effect of edit-
ing events is to make mitochondrial encoded pro-
teins more similar in sequence to their homologous
in other species (Gualberto et al., 1989). For in-
stance, PREPMT (Mower, 2005) and EDIPY (Picardi
and Quagliariello, 2005) are both systems exploiting
this tendency of RNA editing to “correct” codons that
specify unconserved amino acids. A more recent ap-
proach has been proposed in (Lenz and Knoop, 2013).
The simplest way to find genes in a genome is to
scan the nucleotide sequence in all the three possible
reading frames, searching for DNA sequences that do
not contain any stop codon in a given reading frame.
The sequence comprised between a start and a stop
codon is an Open Reading Frame (we call them ORF
sequences in the rest of this paper) and it can be con-
sidered a potential protein encoding segments if its
length is at least 300 nucleotides. The alternative to
this “ab initio” gene discovery is the comparative gene
finding, based on sequence similarity. It consists in
comparing translated sequences with known proteins,
and homology criteria can allow for the identification
of new proteins in the organism under analysis. The
number of known mitochondrial genes varies in dif-
ferent organisms from only 5 genes in Plasmodium
to nearly 100 genes in jakobid flagellates, with the
average across eukaryotes being 40-50 genes (Burger
et al., 2003). Despite the difference in number, mito-
chondrial genes are involved in five basic processes:
invariantly in respiration and/or oxidative phosphory-
lation and translation, and occasionally also in tran-
scription, RNA maturation and protein import. How-
ever, because of the existence of mechanisms increas-
ing gene complexity in plant mitochondria, it is pos-
sible that a certain number of mitochondrial proteins
remains still unknown. Indeed, RNA editing mecha-
nism alters the linearity of genetic information trans-
fer, introducing differences between RNAs and their
coding DNA sequences that hinder both experimental
and computational research of genes. In fact, com-
mon software tools of gene search are helpful in find-
ing canonical genes, but they fail in discovering genes
so encrypted in the genome. Accordingly, complete
sequencing of mtDNA of many organisms allowed the
identification of canonical genes, but much of the in-
formational content of plant mitochondrial genomes
remains still undiscovered. Finding plant mitochon-
drial proteins and understanding how they integrate
into pathways, represent major challenges in cell bi-
ology.
In order to identify new proteins in plant mito-
chondria, we propose a method for ORF sequences
mining from genomes, based on editing simulation,
as illustrated in Section 2. Our approach aims at iden-
tifying ORFs that could potentially be coding regions
for proteins but that, due to RNA editing, cannot be
detected by classical finding techniques. The pre-
sented method is based on the observation that plant
mitochondria use editing mechanism on crucial sites,
for example to generate start codon aug from acg.
The main idea we pursue is that of simulating such an
editing process by exploiting a suitable metric to com-
pute the distance between sequences, in such a way to
directly take editing into accounts. We applied our
method on the mtDNA of Oryza sativa (rice), obtain-
ing encouraging preliminary results that are described
in Section 3. First, our method was able to single
out amino acid sequences corresponding to rice pro-
teins for which start codons editing is known to occur,
whereby validating our approach. Second, a number
of protein sequences were predicted, some of which
are homologous to proteins expressed in other organ-
ims, while some others are completely novel ones.
2 METHODS
The idea exploited in this work is that of trying to
automatically mimic those editing mechanisms pos-
sibly causing the presence of proteins that are not
imputable to ORF sequences obtained by traditional
methods (e.g. ORF FINDER
1
, STARORF
2
). This
is rather meaningful in plants, where mtDNA edit-
ing mechanisms can often involve nucleotide triplets
leading to start and stop codons. Our approach is
based on the simulation of such a process, in order to
generate novel potential proteins, not yet discovered
in a given input organism. The by far most frequent
nucleotide substitution caused by editing is c → u at
the RNA level, that is, c → t if we refer to mtDNA.
Thus we consider only this kind of nucleotide sub-
stitution in our analysis. Since RNA editing might
occur also on portions inside the simulated ORF se-
quences, we handle also a further editing simulation
step. In particular, when an amino acid sequence is
intercepted for a specif organism, a first criterion to
understand its biological relevance is searching for
significant homologies. Thus, we generate those edit-
ing substitutions on the ORF sequences in such a way
that possible new homologies with known proteins of
other organisms can be detected. To this aim, a suit-
able sequence distance measure is considered, and for
1
http://www.ncbi.nlm.nih.gov/projects/gorf/
2
http://web.mit.edu/star/orf/
Discovering New Proteins in Plant Mitochondria by RNA Editing Simulation
183

each ORF sequence, only those editing substitutions
are generated such that a significant homology with
some of the known proteins is reached, thus avoid-
ing an exponential growth of the sequences to ana-
lyze. Finally, in order to understand if the produced
amino acid sequences can be considered indicative of
gene activity, a further filtering step is carried out by
searching for the presence of possible transcripts in
DBEST (Boguski et al., 1993).
Figure 1 graphically illustrates the main steps of
our method and the associated supporting software
tools. Below we explain in detail each specific step
of our prediction approach.
2.1 Editing on the Start/Stop Codons
In order to extract novel ORF sequences from the
genome of a given organism, edited nucleotide triplets
corresponding to the start and stop of an amino acid
sequence have to be intercepted on the DNA se-
quence. Such triplets are called start codons and stop
codons, respectively. Exist one start codon, that is
atg, and three stop codons, that are tag, tga and taa.
Although ORF sequences can be easily searched for
in a genomic sequence by exploiting one of the exist-
ing software tools, such as for example ORF FINDER
and STARORF. These software do not take in account
of editing mechanism. Therefore, in plants, several
proteins are not found from the ORF sequences re-
turned in output by such tools.
To this aim, we start from the mtDNA of a spe-
cific plant, and predict that some editing substitu-
tions might have happened causing the generation of
some start/stop codons. Among all such possible new
codons, only those corresponding to significative po-
tential ORF sequences are taken into account. In par-
ticular, only ORF sequences corresponding to amino
acid sequences of length at least 100 are considered
to correspond to potential proteins. Thus, between a
start and a stop at least 300 nucleotides have to occur
for potential novel ORF sequences to be singled out.
Furthermore, the most frequent nucleotide substitu-
tion caused by editing is c → u at the RNA level, that
is, c → t if we refer to mtDNA. Thus we consider only
this kind of nucleotide substitution in our analysis.
The following example illustrates how new candi-
date ORF sequences can be generated from the origi-
nal nucleotide sequence, by simulating possible edit-
ing substitutions.
Example 1 In Figure 2 a portion of the rice mtDNA
is shown. In particular, in the considered sequence,
there are two stop codons, taa and tag, highlighted by
a widehat. Since no start codon occurs between the
two stops, no candidate ORF sequences would be ex-
tracted without editing simulation. On the contrary,
if we consider possible substitutions c → t leading to
the generation of new codons, then the start codon atg
resulting from the triplet acg in italic can be indeed in-
tercepted. Since between this start codon and the stop
tag there are 102 nucleotide triplets, the subsequence
highlighted in bold, worth considering as a candidate
ORF sequence, can be extracted this way.
The method starts by considering an input nu-
cleotide sequence (in the case we present in this pa-
per, this is the mtDNA of a plant). Such a nucleotide
sequence s
n
is then scanned in all its three possi-
ble reading frames (for both the forward and the re-
verse cases), by considering all the substitutions c → t
that can generate new start/stop codons (we call them
edited codons, while original codons are those al-
ready occurring in s
n
). Then, the nucleotide subse-
quences with minimum length 300 between a start
and a stop codons are extracted, by taking care that
only maximal subsequences are considered. And, in
fact, if several useful start codons occur before a same
stop codon, only the first start codon is considered for
the purpose of extracting the corresponding ORF se-
quence. All the other start codons are translated as the
corresponding amino acid Methionine (M) in the re-
sulting amino acid sequence. This avoids intercepting
all the possible subsequences. For what concerns the
stop codons, the first one after the chosen start c
START
is considered, if such a c
STOP
is an original codon. In
such a case, the so individuated subsequence is dis-
carded if its length is less than 300 bases, and we
look for another c
START
. If, instead, c
STOP
is an edited
stop, it is taken into account only if between c
START
and c
STOP
there are at least 300 nucleotides, otherwise
such an edited stop is discarded, and the next c
STOP
is
searched for, by using the same rule. We avoid this
way subdividing a potentially significative sequence
in several smaller meaningless subsequences.
Figure 3 summarizes the editing ORF simulation
method as described above.
2.2 Editing on the Amino Acid
Sequences
Let P
ORF
be the extracted amino acid sequences set:
a first question is to what extent possible RNA edit-
ings occurring in each sequence of P
ORF
may influ-
ence the prediction process (it is just worth recalling
that the only editing we are focusing on here is the
c → u one). Note that, if we simulate editing on the
sequences in P
ORF
, we should take into account all the
possible c → u editing configurations that might pos-
sibly occur, the number of which is 2
k
, where k is the
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
184
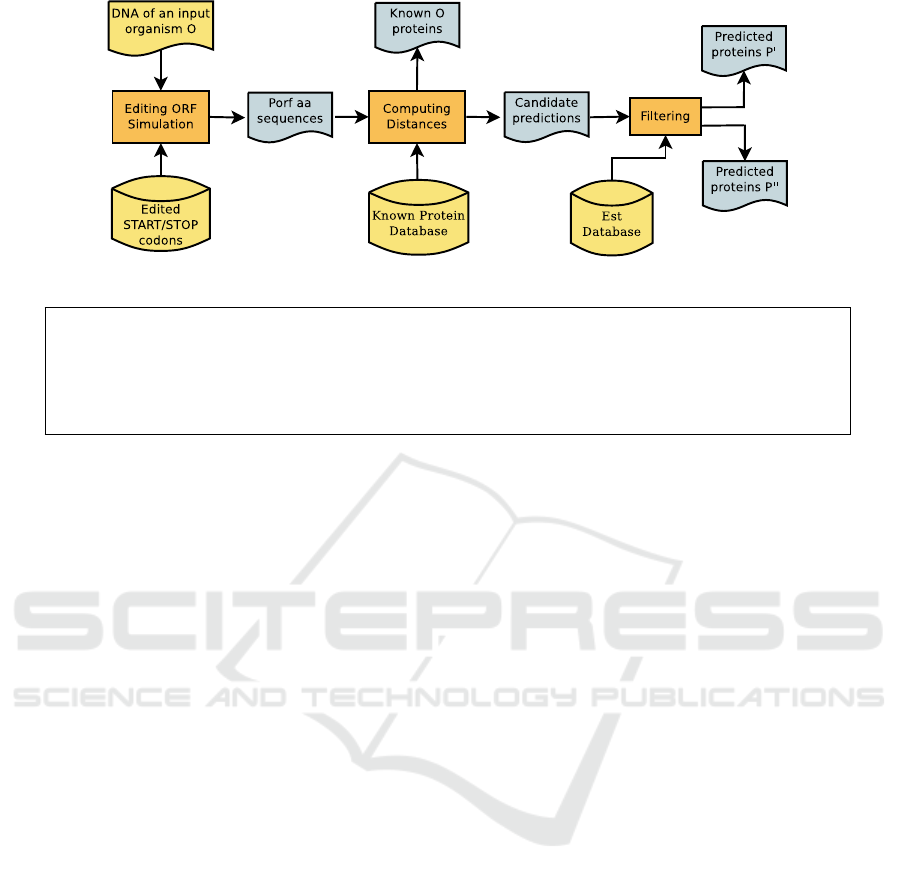
Figure 1: The protein prediction method based on editing simulation.
atc gga tca tca tgc ata atc gaa caa agc tta tcc gca tgg
d
taa agt agt tta cca cac aag tcg aca aaa aag acg ttc ggc
ttt aga aat cat ttt ttt gct ccc tca tcc tcg gtt gtt cgt att tca ttt tct tca aag gca cat gca cta
ggt tac tta cgg aat ctc aaa gaa aga gtc gtc cag gag cac ttc gtt aga ttt gca tgt gtt aag cat ata
gct gaa gtt gcc tat gcg ctt caa cct gct ctt aca aga cga atc tct ttc tat acg caa ttt caa cta gag
tct act cct ttc tgg tct gaa atc tca gta gag acg ata aag att agg tgc ctt tct ttc tat agg gat agg
tgc ttc tct cta
d
tag aaa gaa agg aga tcc agt tta cca ttg aga gta gag aag ggg aag
Figure 2: Editing of the start codon acg → atg.
number of c occurrences in the ORF sequence under
consideration. However, for the purposes of our anal-
ysis, two or more such configurations are to be con-
sidered equivalent as long as they produce the same
amino acid. Note, by the way, that since more than
one c can occur with one single triplet, that triplet
can indeed induce different amino acids via editing
– this is the case, for instance, of the amino acid P
(Proline), that corresponds to four triplets including
ccc and from which, by editing, actually three amino
acids, namely L (Leucine), S (Serine) and F (Pheny-
lalanine), can be obtained. Therefore, a quantitative
analysis is useful here.
Thus, let a
0
be an amino acid containing a c such
that a substitution c → u leads to the generation of
an amino acid a
00
6= a
0
. We say that a
0
is an editable
amino acid. Analogously, we call editable c each c
that may cause the generation of a new amino acid
after a c → u substitution. We exploit the term editing
substitutions to refer to both c → u substitutions and
the corresponding a
0
→ a
00
substitutions, accordingly
to the case under analysis (nucleotide sequences or
amino acid sequences, respectively).
In the following we report an analysis performed
in order to evaluate the effect of editing occurrence
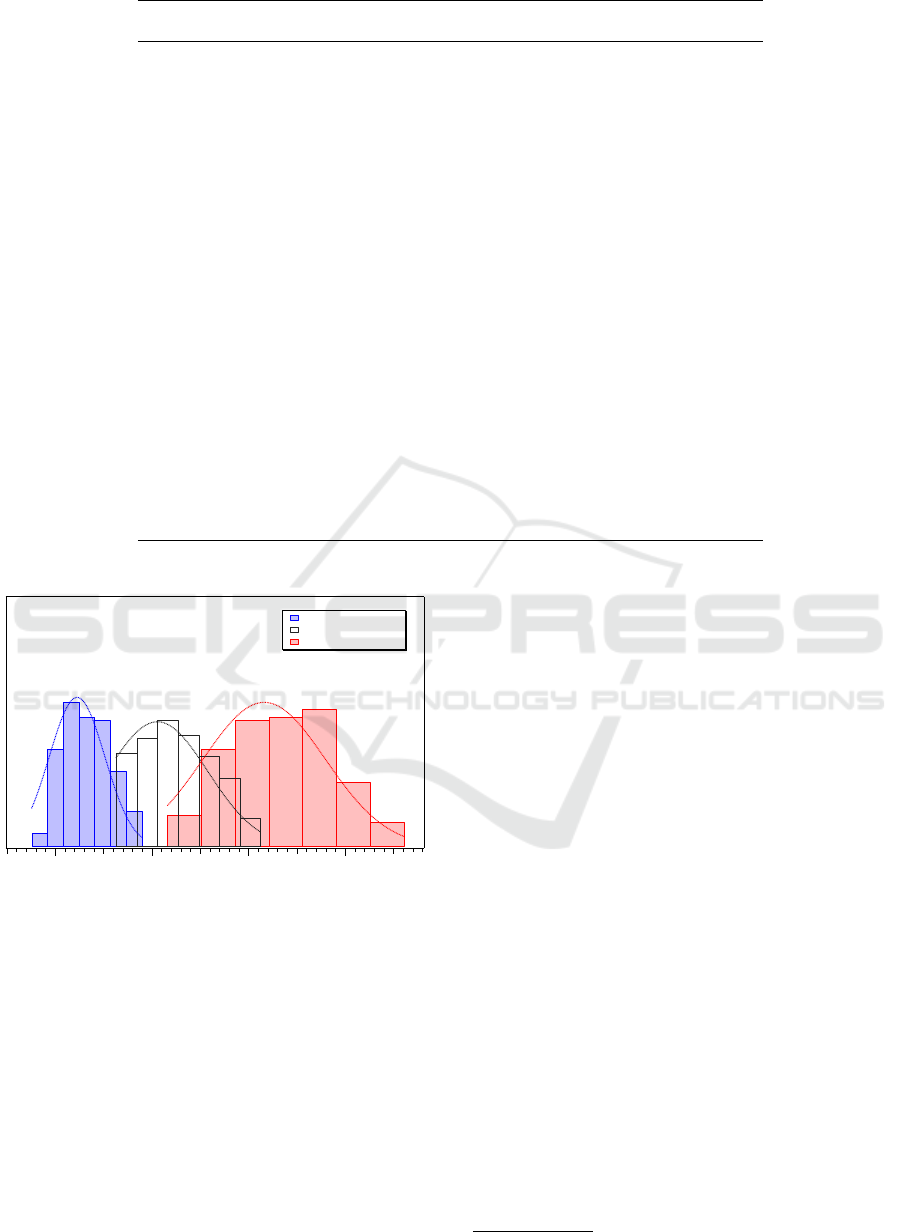
on the amino acid sequences. Figure 4 shows the dis-
tribution of the number of c, editable c and editable
amino acids for unit of length, with respect to all the
amino acid sequences generated from rice mtDNA us-
ing the technique illustrated in the previous section. A
Gaussian fit has been performed for each distribution:
the abscissa corresponding to the peak of each curve
fit has been found to agree with the corresponding cal-
culated average value. Moreover, the expected con-
fidence intervals for normal distributions have been
observed: about 64%, 66%, 67% of the set are within
one standard deviation for fraction of c, editable c and
editable amino acids, respectively. Two standard de-
viations from the mean account for about 98%, 97%
and 95% of the set for each distribution, respectively.
Interestingly, looking at Figure 4, we observe that
the amino acid sequences are more sensible to edit-
ing substitutions than the original candidate ORF se-
quences from which they were obtained. Indeed, the
curve fitting editable amino acids results to be trans-
lated along the x-axis approximatively by a factor 3
with respect to the curve corresponding to editable c.
We also observe that, in some cases, editing substi-
tutions involve more than the 40% of an amino acid
sequence, thus potentially causing also significative
variations with respect to the amino acid sequence
that would have been obtained by translating the orig-
inal nucleotide sequence, without considering editing.
Unfortunately, in order to generate all the differ-
ent amino acid sequences that can be obtained by all
the possible combinations of c → u substitutions, we
should tackle the generation of many possible config-
urations, to be then searched for possible homologies
and/or transcribed sequences. In order to avoid such
a blow-up in the number of candidate ORF sequences
to analyze, we propose the following strategy.
Let s
i
be the amino acid sequence of a candidate
protein, obtained according to the procedure illus-
trated in Section 2.1. We first try to individuate some
known proteins to which s
i
becomes homologous un-
dergoing a suitable editing. The idea is to consider
Discovering New Proteins in Plant Mitochondria by RNA Editing Simulation
185
Input: A nucleotide sequence s
n
;
Output: A set of amino acid sequences P
ORF
;
1. P
ORF
=
/
0:
2. for each of the three possible reading frames fr of s
n
3. repeat
4. repeat
5. read a triplet t from fr;
6. until t is a start codon or by editing t a start codon is achieved;
7. set c
START
to t;
8. repeat
9. read a triplet t from fr;
10. until t is a stop codon or by editing t a stop codon is achieved;
11. set c
STOP
to t;
12. let n
i
be the number of nucleotides between c
START
and c
STOP
;
13. if c
STOP
is an edited stop codon
14. if n
i
< 300
15. skip c
STOP
and goto step 8;
16. end if
17. end if
18. if n
i
≥ 300
19. extract the nucleotide subsequences s
i
between c
START
and c
STOP
;
20. traduce s
i
in an amino acid sequence p
i
;
21. P
ORF
= P
ORF
∪ {p
i
};
22. end if
23. until the end of fr is reached;
24. end for
25. return P
ORF
;
Figure 3: The Editing ORF Simulation Module.
0.40.30.20.1
µ
= 0.12
σ
= 0.03
µ
= 0.32
σ
= 0.06
µ
= 0.21
σ
= 0.05
nr. editable C / length
nr. C / length
nr. editable Aa / nr. aa
Figure 4: Distribution of the number of c, editable c, and
editable amino acids for unit of length in P
ORF
.
a suitable metric to compute the distance between s
i
and each s
j
belonging to a set of known proteins, in
such a way to directly take editing into account. This
way only edited sequences that are homologous to
some already known proteins are generated from s
i
.
In more detail, given a candidate protein with amino
acid sequence s
i
and a known protein with amino acid
sequence s
j
, the distance between s
i
and s
j
is equal
to σ if there exists a set of editing substitutions trans-
forming s
i
into a sequence
e
s
i
, such that the distance
between
e
s
i
and s
j
is σ. We consider significative the
homology between
e
s
i
and s
j
if σ is less than a fixed
threshold σ
th
. In cases where no such an homologous
s
j
can be singled out, we keep the “original” s
i
(indi-
viduated by the Editing ORF Simulation Module) for
further analysis. Otherwise, we choose one among
those
e
s
i
scoring both the lowest σ and the smallest set
of editing substitutions.
We work by minimizing the Levenshtein distance
(Levenshtein, 1966) between sequences, modified to
take into account possible amino acid substitutions, as
shown in the following example.
Protein sequences in P
ORF
for the organisms O
(e.g., Oryza) are compared against known proteins
3
,
by simulating editing as explained above in order to
single out interesting homologies. Some of the se-
quences in P
ORF
can be found to be known O proteins,
in which case we discard them from further analy-
sis. Let P
edited
be the resulting amino acid sequences
set, where the original proteins of P
ORF
are possi-
bly substituted by the edited sequences correspond-
ing to minimum distance configurations. We can di-
vide P
edited
in two further subsets P
0
edited
and P
00
edited
.
P
0
edited
includes amino acid sequences for which sig-
nificant homologies have been found with respect to
some proteins belonging to other organisms, while
P
00
edited
contains the remaining ones.
Figure 5 illustrates the pseudocode for this step of
our approach.
Consider again the ORF sequence discussed in
Example 1. By applying the procedure explained
3
ftp://ftp.ncbi.nlm.nih.gov/blast/db/.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
186

Input: The set of amino acid sequences P
ORF
;
A set of known protein sequences P
known
;
A distance threshold σ
th
;
Output: A set of edited amino acid sequences P
edited
;
1. P
0
edited
, P
00
edited
=
/
0;
2. for each amino acid sequence s
i
∈ P
ORF
3. s
e
, s
h
= ε; /* null string
4. σ
h
= d(s
i
, ε); /* initial distance is set to
the maximum possible value
5. for each protein sequence s
j
∈ P
known
6. find the amino acid sequence
e
s
i
= ϕ(s
i
),
where ϕ is an operator transforming s
i
into
e
s
i
by applying a finite sequence of editing
substitutions to minimize d(
e
s
i
, s
j
);
7. if d(
e
s
i
, s
j
) < σ
h
8. s
e
=
e
s
i
;
9. s
h
= s
j
;
10. σ
h
= d(
e
s
i
, s
j
);
11. end if
12. end for
13. if s
h
does not belong to O and σ
h
≤ σ
th
14. add s
e
to P
0
edited
;
15. if s
h
does not belong to O and σ
h
> σ
th
16. add s
i
to P
00
edited
;
17. end for
18. return P
edited
= P
0
edited
∪ P
00
edited
;
Figure 5: The Computing Distances Module.
in this section, the corresponding amino acid se-
quence, which did not present any significant ho-
mologous without editing, shows high similarity with
V5IJ74 IXORI, a putative atp synthase subunit of the
common tick Ixodes ricinus.
2.3 Final Predictions
The amino acid sequences in P
edited
are further ana-
lyzed by searching for the presence of possible tran-
scripts, since this can be considered indicative of gene
activity. In particular, the DBEST (Boguski et al.,
1993) is queried to this aim by each s
i
∈ P
edited
, in or-
der to detect significant homologies with some known
expressed sequences. Eventually, our system returns
in output two sets of predicted proteins: P
0
and P
00
, re-
spectively containing amino acid sequences in P
0
edited
and in P
00
edited
for which trascripts have been found in
O (e.g., Oryza). As an example, the edited amino acid
sequence of the ORF discussed in Example 1 presents
EST in Zea mays but not in Oryza, thus it has been
discarded.
3 RESULTS
We applied our method on Oryza sativa (rice) mtDNA
with the aim of predicting possible new mitochondrial
proteins. The entire mitochondrial genome of rice has
been sequenced (Notsu et al., 2002); it was found to
be 490,520 bp long. To date, 81 genes have been iden-
tified, 53 of which coding for proteins. The automatic
simulation of editing on all the potential start and stop
codons of rice mtDNA leads to the generation of a to-
tal of 176 candidate ORF sequences, among which
138 are those involving edited start and stop codons.
In order to validate our approach, we at first ver-
ified if the two proteins that are known to be gener-
ated by RNA editing in Oryza sativa were actually
recognized by our system. We found both of them,
the NADH dehydrogenase subunit 1 and the NADH
dehydrogenase subunit 4.
Candidate ORF sequences involving edited start
and/or stop codons consist of 60 sequences with edit-
ing only on the start codon and 78 sequences with
editing only on the stop codons. The latter ones seem
to be less interesting for our analysis, since they repre-
sent subsequences of ORF sequences that can be gen-
erated also by other available ORF finder tools. In
this analysis, we focus only on the former 60 candi-
date ORF sequences. Among them, we found 32 se-
quences corresponding to proteins already described
in rice, 7 not known in Oryza but homologous to pro-
teins identified in other organisms, and 21 sequences
that have been not described before (see Figure 6).
The screening of the DBEST database (Boguski
et al., 1993) by TBLASTN (Altschul et al., 1997)
gave very interesting results: six candidate ORF se-
quences from forward DNA strand and seven from re-
verse strand (Table 1) showed positive matches, indi-
cating their transcription in the organism under study.
Because transcription of an open reading frame indi-
cates gene activity, we directed our further analysis on
these 13 transcribed ORFs. The first column in Table
1 contains progressive numbers indicating the con-
sidered candidate ORF sequences, second and third
columns show the position in the nucleotide sequence
of the start and the stop codons of each sequence, re-
spectively. The last column shows organisms where
the corresponding transcribed ORF has been found.
Among these sequences, five (2 from forward and 3
from reverse strand) were homologous to proteins al-
ready known in other organisms, as reported in Table
2, but eight sequences have never been described until
now. The evidence of RNA transcription from these
sequences let us suppose that they may indeed repre-
sent new genes.
The second and third column in Table 2 show
the query coverage and percent identity of protein
BLAST results, respectively. Among the candidate
ORF showing homology with proteins already known
in other organisms, four are returned by our system
as hypothetical proteins. In particular, sequence 6
Discovering New Proteins in Plant Mitochondria by RNA Editing Simulation
187
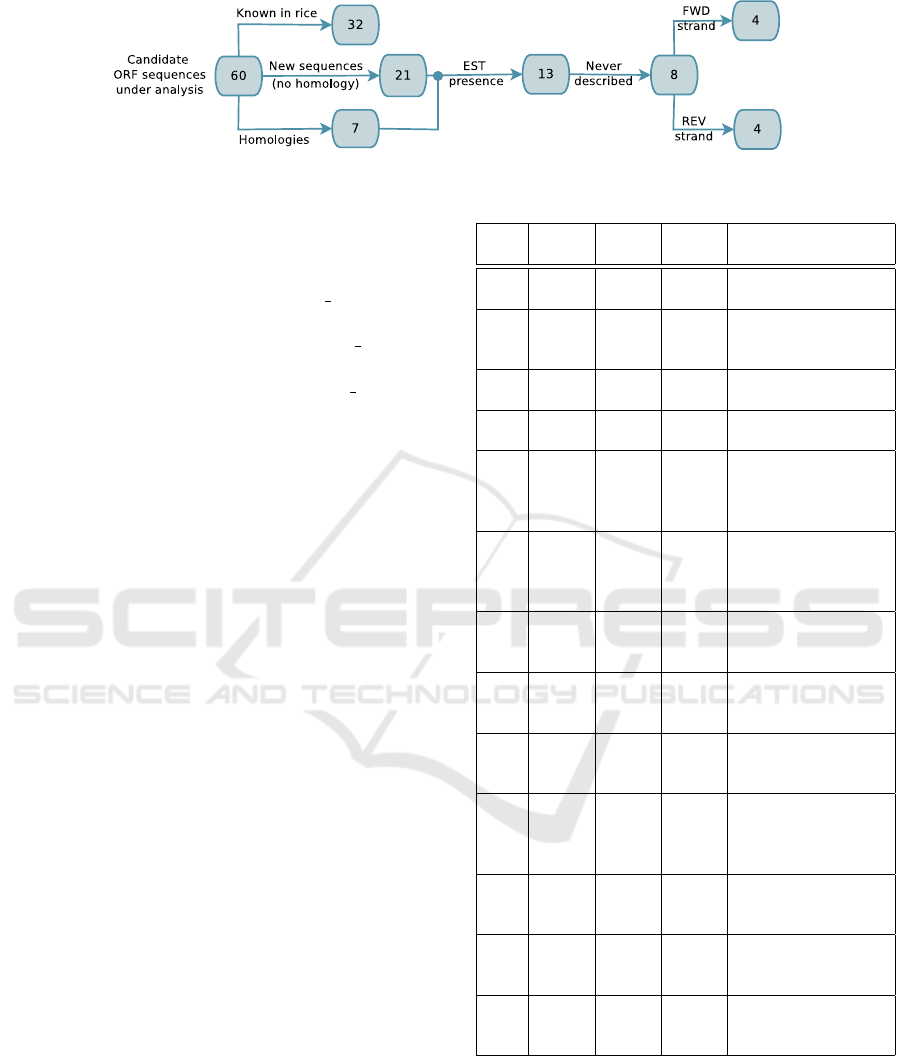
Figure 6: Classification of the discussed sequences.
in Table 2, is homologous to a protein described in
Zea mays (with NCBI accession number AAR91184),
a monocotyledon plant, and in Trichoplax adherens, a
Placozoa. Sequence 7 shows homology with a protein
described in Persephonella marina (Y P 002730925)
and many bacteria, sequence 10 is homologous to a
protein identified in Nicotiana tabacum (Y P 173435)
and other plants, while sequence 12 is homologous to
a protein described in Brassica napus (Y P 717160).
DBEST screening showed that all of them are ex-
pressed not only in Oryza sativa, but in several or-
ganisms. Functional studies can clarify the nature of
these proteins. Sequence 4 showed high similarity
with PG1 protein, a factor involved in transcription
regulation, in several plants and many bacteria. The
high similarity with the same protein in organisms,
even very distant from an evolutionary point of view,
strongly indicates that our candidate ORF sequence
of Oryza actually corresponds to the PG1 protein.
4 CONCLUSION
We proposed a method to predict novel candidate pro-
teins resulting from c → u editing substitutions in
plants mitochondrial DNA. The idea is to simulate the
natural RNA editing mechanism, in order to gener-
ate possible Open Reading Frame sequences coding
for some uncharacterized proteins. The approach al-
lowed us to identify interesting amino acid sequences
in Oryza which could represent proteins yet unknown.
As future work, first of all we will test the method
on the mRNA of other plant mitochondria. Then, we
plan to investigate different strategies for the inner
editing of the candidate sequences, for example based
on the analysis of the context around the c → u substi-
tution (Mulligan et al., 2007). Furthermore, we think
to extend this in order to manage also next genera-
tion sequencing data, as already done in (Picardi and
Pesole, 2013). Finally we observe that, often, pro-
teins with low sequence homology have similar func-
tions and secondary/tertiary structures, whereby it ap-
pears sensible to comparatively look at such struc-
tures for the result assessment purposes, possibly by
suitable prediction techniques (see, e.g., (Palopoli
et al., 2009)).
Table 1: ORF sequences with transcription in rice.
SEQ. QUERY START STOP
EST
NR. COV. COD. COD.
1 124 354085 354460
O. sativa, T. dactiloydes
Z. mays, others
2 108 407800 408127
O. sativa, B. oldhamii
T. dactyloides, Zea,
T. aestivum, S. bicolor
3 99 467635 467935
O. sativa, S. bicolor,
Z. mays
4 111 283844 284180
O. sativa, Z. mays,
several bacteria
5 107 362648 362972
O. sativa, B. oldhamii,
Z. mays, Triticum,
S. bicolor, V. vinifera,
others
6 139 364454 364874
O. sativa, Z. mays,
B. oldhamii, Triticum,
S. bicolor, V. vinifera,
A. thaliana, others
7 200 463889 463286
O. sativa, Z. mays,
V. vinifera, T. aestivum,
others
8 127 232370 231986
O. sativa, B. oldhamii,
Z. mays, T. aestivum,
C. sinensis, others
9 108 449361 449034
O. sativa, Z. mays,
C. papaya, T. dactyloides,
R. communis, others
10 112 314493 314154
O. sativa, Z. mays,
B. oldhamii,
T. dactyloides,
Zea, S. bicolor, others
11 142 295218 294789
O. sativa, E.crassipes,
B. oldhamii,
L. tulipifera, others
12 114 201474 201129
O. sativa, Z. mays,
T. dactyloides,
B. oldhamii, others
13 100 105822 105519
O. sativa, T. aestivum,
Petunia, T. dactyloides,
B. oldhamii, others
ACKNOWLEDGEMENTS
PRIN Project 20122F87B2 “Approcci composizion-
ali per la caratterizzazione e il mining di dati omici”
(toF.F., C.G. and S.E.R.), financed by the Italian Min-
istry of Education, Universities and Research.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
188
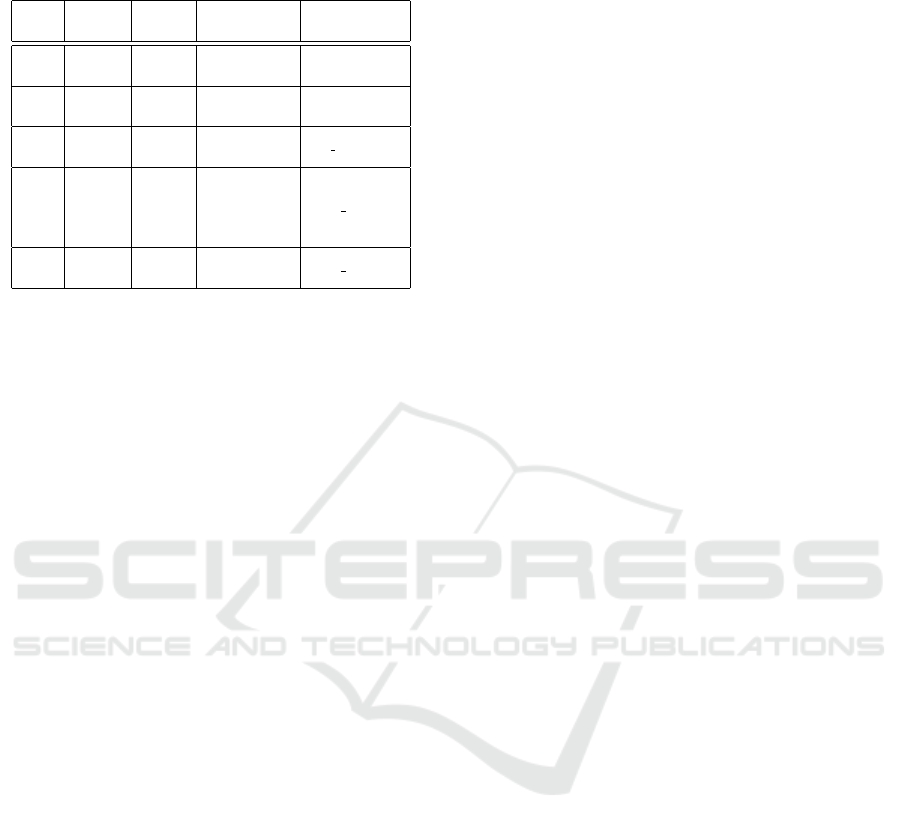
Table 2: ORF sequences with homology to existing proteins
(indicated by their name or NCBI accession number).
SEQ. QUERY IDENT. HOMOLOGUE HOMOLOGUE
NR. COV. ORGANISMS PROTEINS
4 89 49
Some plants,
PG1
many bacteria
6 46 98
Z. mays,
AAR91184
T. ashaerens
7 59 57
P. marina,
YP 002730925
Bacteria
10 95 89
N. tabacum,
YP 173435
B. vulgaris,
A. thaliana,
other
12 69 78 B. napus YP 717160
REFERENCES
Altschul, S. F. et al. (1997). Gapped BLAST and PSI-
BLAST: a new generation of protein database search
programs. Nucleic Acids Research, 25(17):3389–
3402.
Boguski, M. S., Lowe, T. M., and Tolstoshev, C. M. (1993).
dbEST–database for Expressed Sequence Tags. Nat
Genet., pages 332–333.
Brennicke, A., Marchfelder, A., and Binder, S. (1999).
RNA editing. FEMS Microbiol. Rev., 23:297–316.
Bundschuh, R. (2004). Computational prediction of rna
editing sites. Bioinformatics, 20(17):3214–3220.
Burger, G., Gray, M. W., and Lang, B. F. (2003). Mitochon-
drial genomes: anything goes. TRENDS in Genetics,
19(12):709–716.
Gray, M. W., Hanic-Joyce, P. J., and Covello, P. S. (1992).
Transcription, processing and editing in plant mito-
chondria. Annu. Rev. Plant Physiol. Plant Mol. Biol.,
43:145–175.
Gualberto, J. M., Lamattina, L., Bonnard, G., Weil, J. H.,
and Grienenberger, J. M. (1989). RNA editing in
wheat mitochondria results in the conservation of pro-
tein sequences. Nature, 341:660–662.
Hoch, B., Maier, R. M., Appel, K., Igloi, G. L., and Kossel,
H. (1991). Editing of a chloroplast mRNA by creation
of an initiation codon. Nature, 353:178–180.
Lenz, H. and Knoop, V. (2013). PREPACT 2.0: Predicting
C-to-U and U-to-C RNA editing in organelle genome
sequences with multiple references and curated RNA
editing annotation. Bioinform Biol Insights, 7:1–19.
Levenshtein, V. I. (1966). Binary codes capable of correct-
ing deletions, insertions, and reversals. Soviet Physics
Doklady, 10(8):707–710.
Mower, J. P. (2005). PREP-Mt: predictive RNA editor for
plant mitochondrial genes. BMC Bioinformatics, 6:96.
Mulligan, R., Chang, K. L., and Chou, C. C. (2007). Com-
putational analysis of rna editing sites in plant mito-
chondrial genomes reveals similar information con-
tent and a sporadic distribution of editing sites. Mol
Biol Evol, 24(9):1971–1981.
Notsu, Y. et al. (2002). The complete sequence of the rice
(oryza sativa l.) mitochondrial genome: frequent DNA
sequence acquisition and loss during the evolution of
flowering plants. Mol Genet Genomics, 268(4):434–
445.
Palopoli, L., Rombo, S. E., Terracina, G., Tradigo, G., and
Veltri, P. (2009). Improving protein secondary struc-
ture predictions by prediction fusion. Information Fu-
sion, 10(3):217–232.
Picardi, E. and Pesole, G. (2013). REDItools: high-
throughput RNA editing detection made easy. Bioin-
formatics, 29(14):1813–1814.
Picardi, E. and Quagliariello, C. (2005). EdiPy: a re-
source to simulate the evolution of plant mitochon-
drial genes under the RNA editing. Comput. Biol.
Chem., 30(1):77–80.
Picardi, E., Regina, T. M. R., Brennicke, A., and
Quagliariello, C. (2007). Redidb:the rna editing
database. Nucleic Acids Research, 35:D173–D177.
Regina, T. M. R., Lopez, L., Picardi, E., and Quagliariello,
C. (2002). Striking differences in RNA editing re-
quirements to express the rps4 gene in magnolia and
sunflower mitochondria. Gene, 286:33–41.
Schuster, W., Unseld, M., Wissinger, B., and Brennicke,
A. (1990). Ribosomal protein S14 transcripts are
edited in Oenothera mitochondria. Nucleic Acids Res.,
18:229–233.
Takenaka, M., D., D. V., van der Merwe, J. A., Zehrmann,
A., and Brennicke, A. (2008). The process of RNA
editing in plant mitochondria. Mitochondrion, 8:35–
46.
Wintz, H. and Hanson, M. R. (1991). A termination codon
is created by RNA editing in the petunia mitochondrial
atp9 gene transcript. Curr Genet, 19:61–64.
Discovering New Proteins in Plant Mitochondria by RNA Editing Simulation
189
