
PAPAyA: A Highly Scalable Cloud-based Framework for Genomic
Processing
Francois Andry
1
, Nevenka Dimitrova
2
, Alexander Mankovich
2
, Vartika Agrawal
2
,
Anas Bder
3
and Ariel David
3
1
Philips, HISS, 4100 East Third Ave, Foster City, U.S.A.
2
Philips, CISS, 19 Skyline Drive, Hawthorne, U.S.A.
3
Philips, HISS, 18 Aharon Bart St, Petah Tikva, Israel
Keywords: Genomics, Cloud, PaaS, IaaS, Security, Privacy, Asynchronous Processing, Pipelines, Workflow, Big Data,
Analytics, NGS, Oncology, Infectious Diseases.
Abstract: The PAPAyA platform has been designed to ingest, store and process in silico large genomics datasets using
analysis algorithms based on pre-defined knowledge databases with the goal to offer personalized therapy
guidance to physicians in particular for cancers and infectious diseases. This new highly scalable, secure
and extensible framework is deployed on a cloud-based digital health platform that provides generic
provisioning and hosting services, identity and access management, workflow orchestration, device cloud
capabilities, notifications, scheduling, logging, auditing, metering as well as specific patient demographic,
clinical and wellness data services that can be combined with the genomics analytics results.
1 INTRODUCTION
Progress in next-generation sequencing (NGS),
including whole genome and exome sequencing,
transcriptome profiling and detection of single
nucleotide polymorphisms (SNPs), together with
innovations in cloud-based architectures and big-
data analytics stacks is unlocking the power of
genomes towards personalization of healthcare.
There are multiple clinical areas, such as
oncology, infectious diseases, clinical genetics and
others, where genomics can have big impact on
clinical decision-making.
In oncology, it is well known that tumors are
driven by the accumulation of numerous molecular
alterations. In this case, the care for the patient can
be personalized: based on the patient genomic
fingerprint and other sources of information (e.g.
radiology, biopsy, clinical data, family history and
population management data), new therapy options
and clinical trials can be proposed by an oncologist.
When it comes to infectious diseases, the
genomic sequence can help characterize the precise
sub-strains of a particular pathogen and reveal any
inherent resistance it may have to antibiotics. This is
important in molecular epidemiology especially in
the light of the emergence of so-called superbugs
that can spread as hospital acquired infections.
To address these applications, we started with a
prototype: Physician Accessible Personalized
Analytics Application (aka PAPAyA) for research in
breast cancer that relies on multiple genomics
modalities: gene expression and differential DNA
methylation. The application was oriented towards a
patient centric interpretation of microarray data and
biomarker (signature) discovery.
However, with the evolution of genomics and
introduction of next generation sequencing, the
amount of input data increased many-fold and there
was a need for more versatile platform to address
different types of cancer, and allow for building
clinical applications in various disease domains such
as infectious diseases and cardiology.
The initial prototype was to present the concept
of a research platform for biomarker discovery and
decision support in breast cancer (Janevski et al.,
2009).
The focus of the current platform in this paper is
to enable full scalable cloud based architecture and
implementation to support extensibility, reliability,
security and use it for any cancer and any biological
or clinical domain. Another dimension of the new
platform is the versatility to handle any analysis of
next generation sequencing data (expression, copy
198
Andry, F., Dimitrova, N., Mankovich, A., Agrawal, V., Bder, A. and David, A.
PAPAyA: A Highly Scalable Cloud-based Framework for Genomic Processing.
DOI: 10.5220/0005668401980206
In Proceedings of the 9th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2016) - Volume 3: BIOINFORMATICS, pages 198-206
ISBN: 978-989-758-170-0
Copyright
c
2016 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

number variations, methylation, and fusions).
In this paper we describe how the Philips
PAPAyA has been designed to associate genomic
data with clinical data while leveraging the power of
our PaaS: HealthSuite Digital Platform (HSDP) in
order to process genomic data and be able to provide
precision diagnostic, for example, the following
questions:
How to match a tumor’s genotype with a
drug for best outcome?
How to elucidate the cancer subtypes in a set
of RNAseq samples?
How to tackle and prevent the spread of
hospital acquired infections?
However, converting high-throughput genomic
data into clinically actionable information is not a
straightforward task.
The first challenge is to be able to ingest and
store extremely large amounts of genomic data (up
to 1TB for a single patient whole genome) in a
reliable and secure manner while satisfying legal
requirements for long-term storage.
The second challenge is to be able to run
asynchronously parallel processing heterogeneous
pipelines and associated jobs (e.g. sequence
alignment, variant and mutation calling, copy
number variation detection), written in various
programming languages, in a highly quality
controlled, reliable, reproducible and scalable
manner.
The third challenge is to dynamically integrate
domain-specific knowledge from various databases
that may require frequent updates and to generate
clinically actionable results that are reproducible
during subsequent runs.
A fourth difficulty is to manage the potential
users of the platform (researchers, oncologists,
pathologists, physicians, technicians, administrators)
and to create an interactive, role-based and secure
user interface that provides several key functions: to
query the framework; to display the data, metadata
and knowledge-bases used; to define and obtain the
definition of pipelines and modules; to retrieve the
output of the tools; and to generate an intuitive and
comprehensive visualization of reports and
analytics.
There have been many efforts to automate next
generation sequencing data analysis. Galaxy is such
a workflow system that has been used in sequence
analysis and other bioinformatics applications
(Goecks et al., 2012).
Another group developed infrastructure and tools
to support multisite
comparative effectiveness studies using web services
for multivariate statistical estimation in the
SCANNER federated network. (Meeker et al.,
2015).
While these public efforts are excellent vehicles
for research activities, they are not meant for using
in clinical applications.
GenePattern was one of the first systems that
offered to a broad audience a repository of analytic
tools for genomic data (Reich et al., 2006).
Focusing on the web access, Mobyle system
offered a large panel of curated bioinformatics tools
available in a homogeneous environment, to invoke
services distributed over remote Mobyle servers,
thus enabling a federated network of curated
bioinformatics portals without the user having to
learn complex concepts or to install sophisticated
software (Néron et al., 2009).
2 USE CASES
PAPAyA is a framework, deployed on the
HealthSuite Digital Platform, for hosting multiple
genome informatics applications. These applications
inhabit diverse clinical domains and assemble
information from various sources, across many
hospitals and their affiliates either on premise or on
the cloud, and can deliver real-time actionable
information to the clinical experts.
Examples of use cases include breast, prostate
and lung cancer scenarios with oncologists,
pathologists, urologists, and genome informaticists
as users.
3 SYSTEM SPECIFICATIONS
In redesigning the PAPAyA architecture, our team
always had in mind to build a world-class platform
with the following intrinsic properties:
Reliability: Offers dependable mechanisms for
automatic, secure, quality assured acquisition
of demographic, clinical and genomic data
from healthcare organizations. Provide a
reliable way to schedule and execute various
pipelines such as those for detecting variants,
mutations, copy number variation (CNV),
differential gene expression and differential
DNA methylation.
Security: Creates and manages user accounts
with highly secure authentication and
authorization mechanisms, and encrypted data
in flight, but also at rest (including secure
PAPAyA: A Highly Scalable Cloud-based Framework for Genomic Processing
199

Table 1: PAPAyA cancer related usage scenarios.
Patients history/context PAPAyA process/output
Patient is 63 years old,
diagnosed with primary
localized prostate cancer.
Biopsy shows a Gleason
score of 3 (moderately
differentiated carcinoma).
Urologist wants to know
the aggressiveness of the
tumor and options for any
surgical procedure,
chemotherapy or active
surveillance.
Patient's RNA is extracted
from the patient tissue and
PAPAyA runs the RNAseq
pipeline.
Assessment of tumor-driving
cancer signalling pathways
indicates that the patient
does not have an aggressive
disease. The urologist may
suggest a simple follow up
without immediate
intervention.
Patient is a 48-year-old
woman with a distant
history of light smoking
with progressive dyspnea
has been diagnosed with
stage IV lung carcinoma
and has already started
chemotherapy.
However, her condition has
recently deteriorated: fluid
output from a right pleural
catheter, fatigue, weight
loss, hypoxemia, fever and
hypercalcemia.
Oncologist is considering
using a targeted therapy as
an alternative.
Patient RNA transcriptome
sequencing is ordered.
PAPAyA analysed the
sequencing data and whole
exome revealed several
fusions (such as EML4-
ALK). This information
along with radiology and
pathology is presented.
The genomic information
p
oints to efficient drugs
specifically targeting these
fusions (Crizotinib), and the
doctor may propose a few
related clinical trials
targeting the EML4-ALK
fusion.
New renal cell carcinoma
p
atient study is already
underway and the principal
investigator needs to
p
rocess RNAseq data and
exome data.
PAPAyA analysed the
sequencing data using whole
transcriptome detection,
quantification pipeline,
exome alignment and variant
calling.
New renal cell carcinoma
study is underway and the
Principal investigator is
interested in figuring out
which patients have good
p
rognosis and what are the
set of genes (signature) that
stratifies for best outcome.
PAPAyA enabled analysis of
the quantified RNAseq data
to derive a new signature for
p
rognosis of different
subtypes of renal cell tumors
using unsupervised learning
(including deep learning
methods), statistical
evaluation and differential
survival analysis.
Multiple RNAseq studies
are available for study to a
researcher who is looking
for biomarker positive or
signature positive patients
in order to create a clinical
study or enroll patients in a
clinical study.
Cohort analysis within
PAPAyA is conducted with
run-time execution of
“missions” which include
genomic pipelines and
analytics in a cohort
discovery mode, evaluate
classification methods like
random forests for patient
selection (through inverted
matrices).
Table 2: PAPAyA infectious disease scenario.
Patients history/context PAPAyA process/output
More than 50% of patients
in a 26-
p
atient ward of a
p
ublic hospital have been
infected with Methicillin-
resistant Staphylococcus
aureus (MRSA).
The hospital institution
wants to determine the
source of the outbreak.
Swabs are collected from
each patient and sent to
PAPAyA for sequencing the
DNA of the pathogens.
From the resulting genomic
analysis, a phylogenetic tree
of the pathogen SNP data is
derived, to determine
clusters of close genetic
relatedness and a possible
transmission route. Gene
classification for each sub-
sub-strain and associated
drug resistances can help
identify possible treatment.
short- and long-term storage for raw and
analysed genomic data).
Reproducibility: Requires that running a
mission with identical inputs (data and
knowledge-bases) produce the same results. It
must also record all specific versions of
algorithms and databases used for each sample
processed.
Extensibility: Creates workflows for new heath
care domains where genomics can be applied.
Enables the ability to create new mission
cohorts, add new and up-to-date genomic and
medical knowledge bases, as well as create
new access points for advanced research and
clinical trials.
Scalability: Builds new distributed instances of
the pipelines and workflows that can meet
sudden demand increase.
Compliance: Maintains compliance with
HIPAA, HITECH, 21 CFR part 11.
3.1 Data Acquisition
The PAPAyA platform must not only import and
manage new genomic data delivered from NGS
hardware residing in the hospital or at a service lab,
but also patient demographic and clinical data (e.g.
from an EMR), pathology and histological data from
the Laboratory Information Management System
(LIMS) and optionally radiology information from a
Radiology Information System (RIS).
3.1.1 Genomic Data
Genomic input data coming from sequencers (bar-
coded genomic data) will need to be moved to a
database for storing the raw data (FASTQ format),
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
200

processed data (BAM format, VCF format) and all
the metadata associated with the various steps of
library preparation, sequencing and analysis.
Table 3: Genomic data storage requirements.
Data type Raw data Processed
data
whole genome 0.5-1TB 1-2 TB
whole transcriptome 10-20 GB 20-40 GB
targeted
transcriptome
1-4 GB 4-8 GB
targeted,
500 translocations
4-8 GB 8-16 GB
whole exome 20-40 GB 40-80 GB
targeted exome,
500 genes
4-8 GB 8-16GB
3.1.2 Other Patient Data
Demographic and clinical data from EMRs and other
various clinical systems, including pathology and
radiology data from the patient (in HSDP done via a
VHR and an associated EMPI), is imported,
reconciled and associated with biological sequencing
data. We can potentially implement the enterprise
master patient index (EMPI) across multiple
organizations. Our Virtual Health Record (VHR) is
capable of extracting and repurposing data from
multiple different EMR systems (e.g. EPIC, Cerner
and others).
To ensure interoperability, we implement data
standardization aspects following HL7, NAACCR,
CCR, CDA and CCD specifications. We are also
closely involved with GA4GH to keep up with the
various discussions and developments in the
working groups.
3.2 Defining Pipelines and Modules
Pipelines used in PAPAyA are defined as a sequence
of standard jobs (i.e., series of steps embodying
bioinformatics tools and commands) used to
transform data (e.g. sequencing data in FASTQ
format) from a raw state to a processed state in
which various analyses can be performed on variant
calling data in VCF format (Danecek et al. 2011).
Our first pipelines include the steps that help the
creation of phylogenetic tree for infectious diseases
and exome based pipelines for oncology.
Modules used in PAPAyA take this 'usable' data
and apply custom algorithms as well as various
knowledge-base-driven annotations to provide the
user with what is the crux of our platform: providing
clinically actionable information based on a person's
genome or biome.
The flexible design enables incorporation of a new
cancer signature or a new phylogeny building
method to be added.
The modules are implemented using univariate
or multivariate statistical methods as well as various
machine learning and computational biology
algorithms. For example, cancer subtyping can be a
specific module that leverages unsupervised learning
capabilities of the analytics platform.
The distinction that we make is that the role of
modules is to answer specific clinical questions
while pipelines perform computationally intensive
processing that generate intermediate results used as
input to modules themselves.
Certain modules include genomics-oriented
analytics, as well as NLP-related analytics. Our
clinical trial matching module relies upon a well
proven NLP engine that has parsing, tokenizing
capabilities and leverages ontologies such as
SNOMED and LOINC.
3.3 Defining Missions
The highest level of operation within PAPAyA is
accomplished with the concept of “missions”. A
mission is a run time construct that defines several
parameters in a workflow:
Sample or list of samples
Raw, intermediate, or processed genomic data
Pipelines and/or analytics Modules and any
relevant input parameters
Cohort identification
Visualizations of complex genomic data
Querying and Visualizations of integrated
patient and genomic data
The mission is then executed to obtain a desired
output such as performing cohort survival analysis
on a set of patients and returning Kaplan-Meier
curve data. It can also be re-run with identical or
new inputs such as updated pipelines or tools.
A mission can also encompass multiple modules
so that computational biology workflows can be
automated – for example, searching for the most
optimal number of genes that increases the
difference between survival curves of patient
subgroups.
Missions seamlessly enable flexible task
execution, detailed process documentation (for any
retrospective data auditing), and universal
applications for data from infectious disease,
oncology.
PAPAyA: A Highly Scalable Cloud-based Framework for Genomic Processing
201

3.4 Running Asynchronous Jobs
Running a large diversity of genomics tools
associated with pipelines and modules (C, C++,
Java, Perl, Ruby, Python, R) asynchronously in
parallel and at scale requires the platform to be able
to:
provision and host the variety of jobs and their
ecosystems dynamically on demand,
schedule and run these jobs as part of pre-
defined workflows asynchronously,
pass parameters and results that are output
between jobs,
check the status and progress of jobs,
cancel and retry specific jobs,
isolate jobs and associated data for each
specific tenant
3.5 Super Users
PAPAyA provides its users the ability to define
missions and workflows and design their own
pipelines based on the available tools.
It also allows them to monitor the current
processes that are being executed and alerts them to
any errors that may result from substandard input
data.
Figure 1: Super-user design and manage pipelines.
While there are default pipelines and workflows
predesigned in the system, the software enables
users to customize pipelines by mix-and-match of
tools.
3.6 Genomic Knowledge Bases
The final and one of the most important aspects of
genomic analysis is the interpretation of the genomic
results in a functional and/or a clinical context.
For example, when analysing variants for
patients, it is important to identify causal/driver
mutations or clinically actionable variants. In order
to do this, a comprehensive data repository has to be
created that can be used efficiently to annotate
genomic aberrations/alterations (variants, mutations,
CNVs, gene fusions).
This repository is created by integrating various
publicly available data sources such as dbSNP,
Refseq etc. Another important source of clinical
information is the vast pool of literature available on
PubMed.
The entire scientific community is contributing
to this source at an unprecedented level. Using a
blend of natural language processing (NLP) and
manual curation of data can help us extract
meaningful genotype-clinical associations which in-
turn can help make valuable inferences for the
patients’ response/prognosis.
The variant annotation repository of PAPAyA
incorporates all the above-mentioned sources to
annotate aberrations at four levels and thus place the
genomic results in a clinically relevant context.
Figure 2: Knowledge bases for variant annotations.
3.7 Decision Support
Multiple levels of decision support are feasible on
the platform starting with the replacement of the so-
called IVD tests for single genes (for example,
assessment of the mutation status of the BRAF or
the EGFR gene). We have developed the concept of
in-silico assays and the workflows on the platform
are intended to support both clinical practice.
Executing already validated biomarkers
(signatures) as in-silico assays which take
certain cohort patient characteristics,
genomic/transcriptomic data and a decision
function in order to make an inference.
Applying signatures as in-silico assays for
cancer subtyping within an organ based on
classification of tumors as well as pan-cancer.
An example would be breast cancer subtyping
into Luminal A, Luminal B, Her2, and basal
tumors based on a set of 50 genes. Similar
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
202

types of signatures have been used for brain,
ovarian, renal cell carcinoma and others.
Applying predictive signature that can
determine the probability of response to a
certain drug.
3.8 Clinical Research
On the clinical and pharma research side, we
envision support for workflows such as:
Discovery of signatures based on whole
transcriptome data as well as multimodality
signatures leveraging the fact that the platform
has access to genomic, transcriptomic and
methylation data.
Multimodal pathway evaluation in a
neoadjuvant setting to determine the effect of a
certain drug or combination of drugs on the
signalling pathways perturbed by the therapy.
Using genome analytics combined with
machine learning to develop signatures
encompassing stratification/subtyping for best
outcome, prediction to response, prognosis, and
inference of candidates to clinical trials.
4 ARCHITECTURE
The PAPAyA platform is composed of the following
distinct subsystems:
Public Gateway Services: a REST API
exposing all underlying functionalities to be
consumed by PAPAyA front-end application
components.
Genomic Data Persistence: configuration, data
acquisition, read/write operations.
Genomics Pipelines & Modules Processing:
configuration, scheduling, execution and
monitoring.
User Management: user registration (register a
new user account, update profile information,
change and reset password information),
authenticate a user and validate user access
token.
In addition the platform is using the Philips
HealthSuite Digital Platform (HSDP) foundation
services for storage, job execution, user management
and other basic infrastructure functions (Andry et al.,
2015).
4.1 Public Gateway Services
The public gateway services form a thin layer
exposing the public PAPAyA HSDP API for
integration. This layer serves a system entry-point
supplying a client with a public REST interface. The
main logic of the public web services is only to be a
functional secure bridge between public and internal
APIs (data and metadata persistence, configuration,
monitoring, user management).
Figure 3: PAPAyA deployment architecture.
4.2 Genomic Data Persistence Services
The genomic data persistence API service is
responsible for managing genomic data and
associated metadata including:
Create/Update Data: an interface for saving
any genomic data, including raw data and
pipeline intermediate results. The user may
specify additional user-defined metadata for
the file such as batch ID. Asynchronous
uploads of large files is also offered. The user
specifies the location of the data and the
service will copy the data to the persistence and
provide a transaction ID for follow-up.
Read Data: retrieve data from the persistence
according to a specified filter (name, type,
pipeline step). API operations are also
available to check the status of an upload
request. The response will return the state of
the copy and additional metadata.
This internal REST API encapsulates two sets of
services: the genomic data persistence API itself and
an associated configuration API and back-end
façade integration components: genomic metadata
persistence data access object (DAO) and genomics
HSDP storage adapter that decouple the business
PAPAyA: A Highly Scalable Cloud-based Framework for Genomic Processing
203
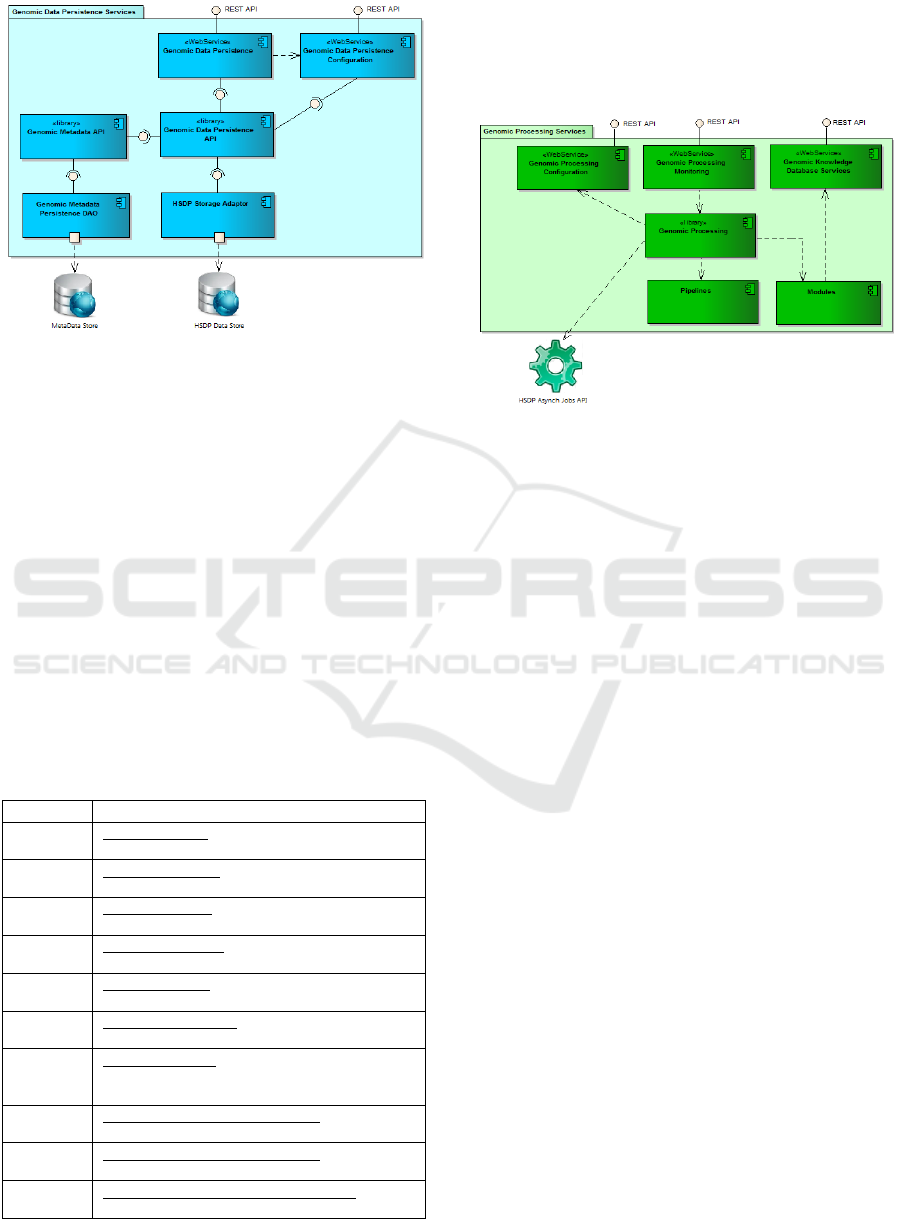
logic of the API and the actual databases.
Figure 4: PAPAyA genomic data persistence services.
4.3 Genomics Processing Module
The genomics processing module is at the heart of
PAPAyA. It includes the following REST APIs:
Genomic processing configuration: to define,
upload, schedule, run or cancel the executions
of missions and associated modules and
pipelines steps and jobs.
Genomic processing monitoring: to get the
status of the execution of missions, modules
and pipelines.
Genomic knowledge database services: to
update or query the genomic knowledge bases
used by the pipelines or the execution of
clinical missions.
Table 4: Genomic processing API operations subset.
Operations description
POST
Create a module
https://<host>/…/modules/{module-id}
GET
Retrieve a module
…/…/modules/{module-id}
POST
Create a pipeline
…/…/pipelines/{pipeline-id}
GET
Retrieve a pipeline
…/…/pipelines/{pipeline-id}
POST
Create a mission
…/…/missions/{mission-id}
GET
Retrieve all missions
…/…/missions
POST
Launch a mission
../../missions/{mission-
id}/sequences/{sequence-id}/executions
DELETE
Cancel the execution of a mission
…/…/executions/{execution-id}
DELETE
Cancel the execution of a mission
…/…/missions/executions/{execution-id}
GET
List all executions of a specific mission
…/…/missions/{missions-id}/executions
The Genomic processing configuration API is the
most complex one since it includes the definition of
the missions and the pipelines steps. Initially, we are
supporting simple sequential pipelines, but in the
future we plan to introduce the ability to create
complex workflows.
Figure 5: PAPAyA processing services.
4.4 User Management
According to the HIPAA regulation, protected health
information (PHI) is individually identifiable health
information including demographic data (HIPAA,
1996), but also genomic information which is very
specific to a particular individual. The disclosure of
genomic data can expose details about health status
and risks, not only for the patient, but for his
relatives as well.
PAPAyA is a tool offered to both clinicians and
researchers. While there is a motivation to increase
the amount of data to be analysed and processed for
clinical trials and research projects, it is imperative
to prevent the accidental re-identification of the data
and leaks of PHI information from cohort(s) of
patients. Frameworks such as GeneCloud (Carey et
al., 2014) used by PAPAyA try to address these
privacy concerns providing stronger security and
privacy guarantees.
The current implementation of PAPAyA is using
the Philips HSDP identity and access management
(IAM) services. This security façade API enables an
application such as PAPAyA to register and log in to
HSDP infrastructure by providing end user
authentication and authorization based on his/her
role (e.g. a researcher does not have the same data
access rights as an oncologist or a system
administrator).
The HSDP IAM services also provide means to
update user profiles, credentials, roles, groups and
token management. HSDP logging and auditing
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
204

services are also used in tracking suspicious data
access transactions, troubleshooting and ensuring
regulatory compliance.
5 DEPLOYMENT
Most of the PAPAyA modules are implemented
using R/Bioconductor, python and Java/Spring and
can be provisioned in a Linux-based container and
deployed in a healthcare organization IT data center
or in a cloud-based environment.
Through the Philips HSDP PaaS layer, it is
possible to provision on-demand access to a shared
pool of configurable and elastic computing resources
(networks, servers, storage, services) based on the
processing needs (Andry et al., 2015).
HSDP runs on top of various types of
infrastructure as a service (IaaS) architectures and
vendors, including private or public clouds, on-
premise, or any hybrid combination.
The fact that PAPAyA is deployed on top of
HSDP makes it very scalable. Through service
brokering and specific REST APIs, PAPAyA micro
services components instance can be scaled up and
down on demand extremely quickly and jobs
associated to missions and pipelines can be
scheduled and run in parallel.
In addition to this, the status of VMS, CPU,
memory usage and storage can be monitored and
trigger notifications if needed so the DevOps and
support teams can make proper adjustments when
necessary.
Through the HSDP asynchronous jobs APIs,
pipeline steps are deployed and run inside Linux
Docker containers, which provide portable, light
footprint runtime requirements and extremely quick
deployment of managed images. As a result, any
genomics tool can be easily integrated as long as
they are initially “cloudified”; in other words, able to
read/write data inputs/outputs from and to a cloud
infrastructure securely and at scale.
6 CONCLUSIONS
Currently, there are 13 million cancer survivors in
the US alone and 1.6 million new cases every year.
Many new therapies are being introduced (Hal,
2013) that require more detailed knowledge of the
genomic makeup of the tumor in order to make a
difference in patient outcome. Some of these new
therapy agents are in the ballpark of $100,000 per
year per patient. While the cost of sequencing has
been decreasing, it is still over $1000 to profile a
tumor and a normal sample.
To match a targeted therapy with a targeted
molecule, a specific genomic candidate aberration
must be identified. Many of these aberrations are
statistically rare, their real direct effect is unknown,
and even after matching, only certain subpopulations
of patients may have better outcome. Our platform is
capable of learning from each one of these outcomes
to put this knowledge back in the service of
practicing clinicians.
Our PAPAyA platform for genome analytics,
and execution of validated tests, can be used for
decision support in clinical practice only when we
instantiate modules that perform assessment of
genomic aberrations with known clinical
significance. In addition, it can serve as the
continuous learning platform for cancer research to
provide unique value to patient care by deriving
more tailored therapy plans, aiding in decisions
around aggressiveness of treatment approach, and
recommending patients to clinical trials.
As part of the planned extensions of the
PAPAyA platform, we would like not only to
provide pre-defined workflows for the missions and
pipelines via an API, but also the ability for certain
advanced users to define these workflows through
visual graphic intuitive standards tools, including
support for Business Process Model and Notation
(BPMN 2.0) processes definition.
ACKNOWLEDGEMENTS
We are very grateful to Patrick Cheung, Yong Mao,
Konstantin Volyanskyy, Mine Danisman-Tasar, who
have been contributing to this project. Thank you to
the PAPAyA R & D team in New York who made
this project possible over many years of hard work
especially Nilanjana Banerjee, A. Janevski, Vinay
Varadan, Sitharthan Kamalakaran. Thank you to the
Philips PAPAyA and the hybrid storage teams in
Petah Tikva, Israel for help in making PAPAyA a
full commercial product. We thank Chad Evans and
the HSDP DevOps team for their contribution on
asynchronous processing. We are also very grateful
to the Iron.io team, especially Chad Arimura for
their help with the IronWorker stack.
PAPAyA: A Highly Scalable Cloud-based Framework for Genomic Processing
205

REFERENCES
Andry, F., Ridolfo, R., Huffman J., 2015, Migrating
Healthcare Applications to the Cloud through
Containerization and Service Brokering, 8th
International Conference on Health Informatics
(HealthINF 2015), pp. 164-171, Lisbon, Portugal.
BPMN 2.0, Business Process Model and Notation V2.0,
2011, http://www.omg.org/spec/BPMN/2.0/.
Carey K., Dimitrova, N., Grantham B., Agrawal, V.,
Nilsson, J., Krasinski, R., 2014 Securing Genomic
Computations for Research and Clinical Decision
Support, 1st PETS Workshop on Genome Privacy
(GenoPri), Privacy Enhancing Technologies
Symposium Amsterdam, NL 2014.
Danecek P., Auton, A., Abecasis, Albers, C., Banks, E.,
DePristo, M., Handsaker, R., Lunter, G., Marth, G.,
Sherry, S., McVean, G., Durbin, R., The variant call
format and VCFtools, Bioinformatics, 27(15):2156-
2158. Genome Privacy (GenoPri), Privacy Enhancing
Technologies Symposium, Amsterdam, NL.
Goecks J., Nekrutenko, A., Taylor J., 2012, Lessons
learned from Galaxy, a Web-based platform for high-
throughput genomic analyses, in Proceedings of the
2012 IEEE 8th International Conference on E-Science
(e-Science), IEEE Computer Society.
Hall, S., 2013, The Cost of Living, New Yorker Magazine,
http://nymag.com/news/features/cancer-drugs-2013-
10/.
HIPAA, Health Insurance Portability and Accountability
Act of 1996 (HIPAA) Privacy and Security Rules,
http://www.hhs.gov/ocr/privacy/.
Hull, D., Wolstencroft, K., Stevens, R., Goble, C., Pocock,
M., Li, P., Oinn, P., 2006, Taverna: a tool for building
and running workflows of services, in Nucleic Acids
Research, Vol. 34, Web Server issue W729–W732.
Janevski, A., Kamalakaran, S., Banerjee, N., Varadan, V.,
Dimitrova, N., 2009, PAPAyA: a platform for breast
cancer biomarker signature discovery, evaluation and
assessment, BMC Bioinformatics, 10 (Suppl 9): S7.
Meeker, D., Jiang, X., Matheny, M., Farcas, C., D'Arcy,
M., Pearlman, L., Nookala, L., Day, M., Kim, K.,
Kim, H., Boxwala, A., El-Kareh, R., Kuo, G., Resnic,
F., Kesselman, C., Ohno-Machado, L., 2015, A
System to Build Distributed Multivariate Models and
Manage Disparate Data Sharing Policies:
Implementation in the Scalable National Network for
Effectiveness Research in the Journal of the American
Medical Informatics Association, Jul 3 2015.
Néron, B., Ménager, H., Maufrais, C.,Joly N., Maupetit, J.,
Letort, S., Carrere, S., Tuffery P., Letondal C., 2009,
Mobyle: a new full web bioinformatics framework,
Bioinformatics, 25:3005-3011.
Reich, M., Liefeld. T., Gould, J., Lerner, J., Tamayo, P.,
Mesirov, J., 2006, GenePattern 2.0., in Nature
Genetics, 38:500-501.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
206
