
Inference of Predictive Phospho-regulatory Networks from LC-MS/MS
Phosphoproteomics Data
Sebastian Vlaic
1
, Robert Altwasser
1
, Peter Kupfer
1
, Carol L. Nilsson
2
, Mark Emmett
3
,
Anke Meyer-Baese
3
and Reinhard Guthke
1
1
Leibniz Institute for Natural Product Research and Infection Biology, Hans-Kn¨oll-Institute,
Beutenbergstr. 11a,D-07745 Jena, Germany
2
Department of Pharmacology & Toxicology, The University of Texas Medical Branch,
Galveston, Texas 77555-1074, U.S.A.
3
Department of Scientific Computing, Florida State University, Tallahassee, Florida 32310-4120, U.S.A.
Keywords:
Phospho-regulatory networks, Network Inference, Glioblastoma Cancer Stem Cells.
Abstract:
In the field of transcriptomics data the automated inference of predictive gene regulatory networks from high-
throughput data is a common approach for the identification of novel genes with potential therapeutic value.
Sophisticated methods have been developed that extensively make use of diverse sources of prior-knowledge
to obtain biologically relevant hypotheses. Transferring such concepts to the field of phosphoproteomics data
has the potential to reveal new insights into phosphorylation-related signaling mechanisms. In this study we
conceptually adapt the TILAR network inference algorithm for the inference of a phospho-regulatory network.
Therefore, we use published phosphoproteomics data of WP1193 treated and IL6-stimulated glioblastoma
stem cells under normoxic and hypoxic condition. Peptides corresponding to 21 differentially phosphorylated
proteins were used for network inference. Topological analysis of the phospho-regulatory network suggests
lamin B2 (LMNB2) and spectrin, beta, non-erythrocytic 1 (SPTBN1) as potential hub-proteins associated with
the alteration of phosphorylation under the observed conditions. Altogether, our results show that inference
of phospho-regulatory networks can aid in the understanding of complex molecular mechanisms and cellular
processes of biological systems.
1 INTRODUCTION
Network inference is a well established tool to un-
cover the complex underlying regulatory mechanisms
of biological systems. The immediate reaction of a
cell towards changing conditions or external perturba-
tions is often mediated by alteration of already exist-
ing proteins via post-translational modifications such
as phosphorylation, methylation, acetylation, sumoy-
lation or ubiquitination (Seet et al., 2006).
Changes in phosphorylation were found to have
an impact on almost all aspects of cell biology
(Jørgensen and Linding, 2008). Therefore, the analy-
sis of causal relations between changes in phosphory-
lation state of proteins is essential for understanding
regulatory mechanisms of the biological system stud-
ied. For the inference of gene regulatory networks
(GRNs) a variety of diverse approaches have been
proposed, which can be used to identify causal rela-
tions in the expression of genes (Hecker et al., 2009b).
The adaptation and application of concepts developed
for GRNs to data from other layers of regulation has
therefore the potential to provide new insights into
regulatory relations of cellular processes.
With analytical methods such as high resolution mass
spectrometry, the quality and quantity of phosphopro-
teomics data is continuously increasing. This offers
new possibilities but also challenges regarding inter-
pretation and computational analysis of large scale
data sets. (Terfve and Saez-Rodriguez, 2012) di-
vided the approaches that have been applied to high-
throughput phosphoproteomics data into two cate-
gories. On the one hand, there are descriptive meth-
ods such as mapping of measured data to pathways,
enrichment analysis, and supervised as well as unsu-
pervised learning methods. These approaches pro-
vide an overview of the data and often serve as a
starting point for a more detailed investigation. On
the other hand, methods such as difference and dif-
ferential equations, Bayesian, boolean and rule-based
Vlaic, S., Altwasser, R., Kupfer, P., Nilsson, C., Emmett, M., Meyer-Baese, A. and Guthke, R.
Inference of Predictive Phospho-regulatory Networks from LC-MS/MS Phosphoproteomics Data.
DOI: 10.5220/0005743000850091
In Proceedings of the 9th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2016) - Volume 3: BIOINFORMATICS, pages 85-91
ISBN: 978-989-758-170-0
Copyright
c
2016 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
85

networks are considered to be predictive since they
are able to process measured data and produce inter-
pretable models that predict the behavior of biological
systems.
In this study, we apply an inference algorithm that
combines these two concepts by using knowledge of
descriptive approaches to produce predictive models,
based on a system of linear equations. To this end,
we apply the previously published transcription fac-
tor binding site integrating LARS (TILAR) algorithm
(Hecker et al., 2009a), which uses a linear model to
infer GRNs from expression data based on least an-
gle regression (LARS) (Efron et al., 2004). One fea-
ture of TILAR is its ability to make use of several
sources of prior-knowledge. Initially, a network struc-
ture template is created, which is iteratively refined to
explain the measured data in the best possible way,
while integrating as much prior-knowledge as possi-
ble. The concept of modeling is based on the idea that
the expression of genes is regulated by transcription
factors (TFs). This introduces the possibility of using
known transcription factor binding sites (TFBSs) as
knowledge base to group genes that are regulated by
the same TFs.
Analogous to the TFBS-based concept of TILAR, we
will show how prior-knowledge about phosphoryla-
tion regulating phosphatases and kinases (PKs) can
be used to group different phosphorylation sites creat-
ing a network template for the inference of phospho-
regulatory networks (PRNs). We will exemplify this
idea by applying the algorithm to published phospho-
proteomics data of glioblastoma stem cells (GSC11)
perturbed with the JAK2/STAT3 phosphorylation in-
hibitor WP1193, IL-6 stimulation and hypoxia.
2 METHODS
2.1 Data Pre-processing
Raw data from (Nilsson et al., 2010) was processed
using R 2.14 (R Core Team, 2014). Duplicated pep-
tide measurements were mean averaged and log10
transformed. Peptides containing no tyrosine, serine
or threonine amino acid were removed from the list of
measured peptides, since only those can be phospho-
rylated.
2.2 TILAR Concept of Modeling for the
Inference of GRNs
The basic idea of the TILAR algorithm as published
by (Hecker et al., 2009a) is to include knowledge
about the expression regulation of the modeled genes
by TFs. Accordingly, the resulting GRNs are com-
posed of relations between the regulating TFs and
their target genes (TF-to-gene relations) as well as re-
lations between the modeled genes and the regulating
TFs (gene-to-TF relations). The TF-to-gene relations
can be, e.g., derived from literature, TFBS-databases
or TFBS prediction tools and serve as a network struc-
ture template for the inference of the gene-to-TF re-
lations. The prior-knowledge structure template can
thus effectively assist the algorithm in the identifi-
cation of biologically meaningful networks. Using
TILAR, the predicted expression value of a gene i (ˆx
i
)
(with i = 1. . . N) is the sum of the weighted expres-
sion values (w
kj
x
j
) of all other genes j via the TF
k (with k = 1. . . F) if (1) the regulated gene i has a
TFBS for the TF k and (2) the gene j does not have a
TFBS for the TF k (equation 1).
ˆx
i
=
F
∑
k=1
N
∑
j=1
(1− b
kj
)w
kj
x
j
b
ki
b
kj
=
1, if gene j possesses a binding
site for TF k
0, else
(1)
The result is a system of linear equations for each
gene that has a TFBS for at least one TF. The inter-
action weights w
kj
corresponding to the gene-to-TF
relations can then be estimated by LARS regression
(see (Hecker et al., 2009a) for details).
2.3 Network Inference
Network inference of PRNs is performed similar to
the inference of GRNs using TILAR (see (Hecker
et al., 2009a) for details). The conceptually adapted
TILAR algorithm uses the PK binding knowledge as
a structure template. Variable selection and estima-
tion is performed using LARS. Initially, the model
that minimizes Mallows Cp statistic (Mallows, 1973)
is selected and the achieved residual sums of squares
(RSS) serves as the initial error. Subsequently, step-
wise backward elimination is performed to iteratively
remove PK-to-peptide edges from the template for
which inference with LARS results in a model with a
RSS lower than the one measured in the previous iter-
ation. The iterative algorithm stops when the removal
of a PK-to-peptide knowledge edge does not decrease
the RSS of the model. The final model is then selected
based on the drop of the RSS compared to the amount
of model parameters. The model parameters are then
estimated using linear regression.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
86

3 RESULTS
3.1 Data Pre-processing
Peptides with an absolute log10 fold-change greater
0.3 between treatment and control were selected as
differentially phosphorylated peptides (DPPs). This
was checked for the WP1193 treated, IL6 stimulated
cells with respect to their corresponding WP1193
treated control under normoxic (NO) (IL6-NO) and
under hypoxic (HO) (IL6-HO) conditions separately.
11 DPPs were identified for the IL6-NO condition and
13 DPPs for the IL6-HO condition, each correspond-
ing to a unique protein (Table 1). The results show
that only 2 peptides overlap between these two sets of
DPPs, corresponding to septin 2 (SEPT2) and spec-
trin repeat containing, nuclear envelope 2 (SYNE2).
Furthermore, we identified 28 DPPs between the two
stimuli, i.e., IL6-NO and IL6-HO respectively (HO-
NO), to include the effect of the condition under
which the cells where cultured.
Table 1: Proteins (Name) corresponding to the DPPs iden-
tified in the different comparisons.
Comparison Name
IL6 - NO EIF3C, GTF2F1, LMNA, MSN, NES,
NUMA1, SEPT2, SLTM, SPTBN1,
SRSF1, SYNE2
IL6 - HO EIF4G3, FSCN1, HSP90B1, ILF3,
KRT1, LMNB2, MARCKS, NOLC1,
RBM8A, SEPT2, SYNE2, TUBB3,
YBX1
HO - NO CLASP1, EIF3C, EIF4G3, G3BP1,
GTF2F1, ILF3, KRT1, LMNB2,
MAP1B, MARCKS, MCM3, MSN,
MYH9, NES, NOLC1, NUMA1,
PI4KB, PLCB3, SEPT2, SEPT7,
SLTM, SRSF1, SRSF11, SYNE2,
TUBB3, VIM, YBX1
DAVID (Huang et al., 2009) was applied to identify
significantly enriched Gene Ontology (GO)-terms us-
ing the union of all sets of DPPs. For a Benjamini-
Hochberg (BH)-corrected p-value < 0.01 the results
show mainly categories related to the cell cycle or
structure building processes (Table 2).
3.2 Network Inference
3.2.1 Application of the TILAR Concept to
Phosphoproteomic Data
The TILAR algorithm is primarily applied for the
inference of GRNs using gene expression data (see
methods). The basic idea of the adapted algorithm is
that a change in the phosphorylation state of a peptide
Table 2: List of significantly enriched GO-terms identified
with DAVID. Shown are the GO-terms (GO-term), the cor-
responding id (GO-term ID), the number of DPPs associ-
ated with the term (Count) and the BH-corrected p-value
(Pvalue).
GO-term
ID
GO-term Count Pvalue
GO:0005198 structural molecule
activity
12 1.16e-5
GO:0008092 cytoskeletal protein
binding
9 8.47e-4
GO:0000279 M phase 8 1.56e-3
GO:0003779 actin binding 7 3.35e-3
GO:0022403 cell cycle phase 8 3.51e-3
GO:0003723 RNA binding 9 5.15e-3
GO:0007049 cell cycle 9 7.77e-3
GO:0048285 organelle fission 6 8.23e-3
GO:0022402 cell cycle process 8 8.46e-3
GO:0000087 M phase of mitotic
cell cycle
6 9.26e-3
alters the activity of the corresponding protein. As a
result, this protein potentially changes the activity of
PKs and therefore, the phosphorylation state of an-
other peptide. Given our data the identified DPPs all
correspond to a unique protein and thus, the adaption
is straight forward and can be formulated as outlined
in equation 1 in the methods section. The predicted
change in phosphorylation state of the peptide i (x
i
, with i = 1. . . N) is the sum of the weighted (w
kj
)
change in activity of the protein j (w
kj
x
j
) via the PK
k (with k = 1. . . F ) if (1) the change in the phosphory-
lation state of peptide i is mediated by the PK k , and
(2) the phosphorylation state of any peptide of protein
j is not.
3.3 Extraction of Prior-knowledge
According to the adapted concept there are two types
of prior-knowledge that can be integrated into the in-
ference process, i.e., knowledge about PKs that are
known to alter the phosphorylationof any of the DPPs
(PK-to-peptide) and knowledge about relations be-
tween the corresponding proteins and the regulating
PKs (protein-to-PK). Accordingly, PhosphoSitePlus
(Hornbeck et al., 2012) was used to find PKs known
to alter the phosphorylation state of the DPPs. Using
this approach, regulators for five DPPs were extracted
correspondingto the proteins phospholipaseC, beta 3,
phosphatidylinositol-specific (PLCB3), minichromo-
some maintenance complex component 3 (MCM3),
myosin (MSN), heavy chain 9, non-muscle (MYH9),
SEPT2 and eukaryotic translation initiation factor
4 gamma, 3 (EIF4G3). Additionally, NetworKIN
(Linding et al., 2008) as well as GPS 2.0 (Xue et al.,
2008) were used to predict binding PKs for the re-
Inference of Predictive Phospho-regulatory Networks from LC-MS/MS Phosphoproteomics Data
87
maining DPPs. The obtained prior-knowledge net-
work was used as a template for the network infer-
ence with TILAR. We also used PathwayStudio 9
(Nikitin et al., 2003) to find known regulatory interac-
tions between the proteins of the corresponding pep-
tides and the identified PKs (protein-to-PK) as well
as known regulatory interactions between the proteins
themselves. However, we did not find any supporting
known relation.
3.4 Network Inference
DPPs with corresponding proteins associated to
the GO-terms ’cell cycle’ (9 DPPs) and ’structural
molecule activity’ (12 DPPs) were selected to in-
vestigate the effect of IL6 stimulation in WP1193
treated GSC11 cells with respect to the different oxi-
dation environments. Four DPPs were found to over-
lap between both groups leading to a total of 17
DPPs. While ’structural molecule activity’ is the
term with the smallest p-value we selected ’cell cy-
cle’ as a second category since it is the most gen-
eral term among the other cell cycle related GO-
terms. Moreover, all DPPs annotated with one of the
cell-cycle associated GO-terms are contained in GO-
term ’cell cycle’. Additionally, we included myristoy-
lated alanine-rich protein kinase C substrate (MAR-
CKS), GTPase activating protein SH3 domain Bind-
ing Protein 1 (G3BP1), general transcription factor
IIF, polypeptide 1, 74kDa (GTF2F1) and Y box bind-
ing protein 1 (YBX1), as the corresponding peptides
share common PK regulators with the 17 selected
peptides. Altogether, 21 DPPs were selected for the
inference. After refinement of the initial structure
template and LARS regression (see methods) the fi-
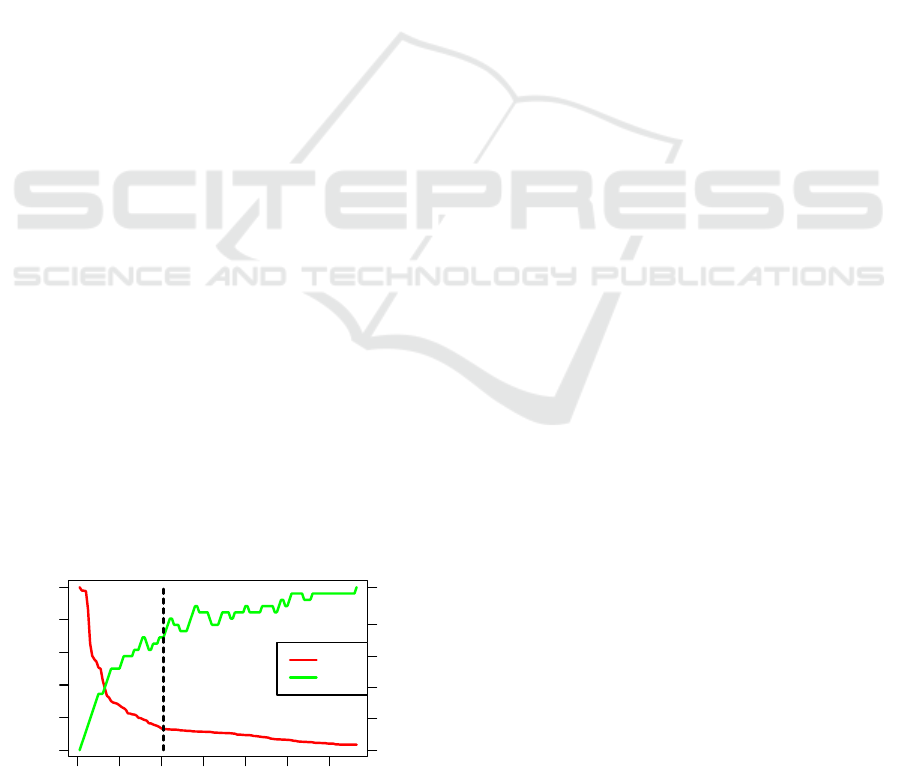
nal model was manually selected. Figure 1 shows that
an increase in the number of parameters (green line)
in the model beyond the selected cutoff of 41 (black
dotted line) does not drastically decrease the RSS (red
line).
The final PRN (Figure 2) is composed of 18 inferred
edges, 36 prior-knowledge edges, 19 PK-nodes and
0 20 40 60 80 100 120
0.0
0.2
0.4
0.6
0.8
1.0
model
RSS scaled to maximum
0
5
10
15
20
26
RSS
size
number of parameters
Figure 1: Drop of the RSS with increasing model size.
18 proteins, representing the corresponding DPPs.
The proteins keratin 1, type II (KRT1), MSN and
YBX1 are not included in the network since no
protein-to-PK edge was inferred connecting the cor-
responding DPPs to any PK in the network. Lamin
B2 (LMNB2) and spectrin, beta, non-erythrocytic 1
(SPTBN1) are the two nodes with the highest out-
degree indicating a signal distributing function in the
PRN. Stimulating the cells with IL6, the peptide that
denotes for LMNB2 is differentially phosphorylated
under HO condition and not affected under NO con-
dition, whereas SPTBN1 is dephosphorylated under
NO conditions and not affected under HO conditions.
Notably, the PRN itself shows a clustering of proteins
associated with either of the two GO terms. Inter-
leukin enhancer binding factor 3, 90kDa (ILF3), nu-
cleolar and coiled-body phosphoprotein 1 (NOLC1),
septin 7 (SEPT7), SEPT2, MARCKS and CLASP1
form a cluster that is mainly regulated by LMNB2
and tubulin, beta 3 class III (TUBB3). These pro-
teins are mostly associated with the mitotic phase of
the cell cycle. Interestingly, all DPPs beside SEPT2
are affected by IL6 treatment in HO, but not NO. In
contrast, GTF2F1, lamin A/C (LMNA), nestin (NES)
and nuclear mitotic apparatus protein 1 (NUMA1) are
mainly regulated by SPTBN1. All of the associated
DPPs are affected by IL6 treatment in NO but not HO.
4 DISCUSSION
In our data set we identified only DPPs correspond-
ing to unique proteins. However, it is well known
that multiple phosphorylation sites can belong to the
same protein and can be phosphorylated indepen-
dently producing many phosphoisoforms leading to
a potentially distinct activity of the protein (Yang,
2005). In cases of multiple DPPs for one pro-
tein, these biological properties are important and
will have to be considered in future studies. Our
results highlight two structurally connected groups
of proteins in the inferred PRN, which are either
mostly differentially phosphorylated in HO (ILF3,
TUBB3, NOLC1, SEPT2, CLASP1, LMNB2) or in
NO (SPTBN1, NES, LMNA, GTF2F1, NUMA1).
Within the PRN the de-phosphorylation of ILF3 and
NOLC1 is mainly due to the negative relation to the
phosphorylated MARCKS. This is in agreement with
literature as ILF3 and NOLC1 are known to be phos-
phorylated during the mitotic phase of the cell cy-
cle (Smith and Miskimins, 2011; Pai et al., 1995).
Moreover, Rombouts et al. hypothesized based on
their data that phosphorylation of MARCKS could in-
hibit the cell cycle (Rombouts et al., 2012). In our
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
88

Figure 2: The inferred PRN. Rectangles denote for PKs and circles represent Proteins of the corresponding DPPs. GO-terms
are outlined as octagons and associated proteins are connected with dotted lines. Blue edges correspond to the extracted
PK-to-peptide prior-knowledge while red and green edges represent the inferred inhibiting and activating edges, respectively.
Colored annotation boxes denote for direction of regulation (green: up; red: down; yellow: none) for the comparisons (left:)
IL6-NO; (middle:) IL6-HO; (right:) HO-NO.
PRN, the increase in phosphorylated MARCKS fol-
lows an increase in LMNB2 phosphorylation. Lamins
are known to be phosphorylated during mitosis to me-
diate matrix disassembly (Dechat et al., 2008). This
indicates that the mitotic phase might be initiated un-
der the HO condition, but not the NO condition, with
MARCKS preventing the phosphorylation of mitosis
important proteins such as ILF3 and NOLC1 as well
as SEPT2 and SEPT7. Both proteins are associated
with the coordination of cytokinesis (Spiliotis et al.,
2005). According to the inferred PRN the second
hub-protein is SPTBN1. In erythrocytes, the phos-
phorylation of this protein was found to be linked
to mechanical stability of the membrane. Manno et
al. demonstrated that increased phosphorylation of
SPTBN1 destabilizes the membrane while reduced
phosphorylation has the adverse effect (Manno et al.,
1995). In the PRN, SPTBN1-phosphorylationis posi-
tively associated with SEPT7 and SEPT2 phosphory-
lation and negatively related to MARCKS phospho-
rylation. This indicates that destabilization of the
membrane by SPTBN1 could be directly linked to
mitosis. The data shows that SPTBN1 phosphory-
lation decreases upon IL6 stimulation in NO while
no effect is seen in HO. Other proteins connected to
SPTBN1 are NUMA1, NES, GTF2F1 and LMNA, all
of which are negatively related to SPTBN1 phospho-
rylation. NUMA1 was reported an important mitotic
component by contributing to the initiation and main-
tenance of the focused spindle poles (Sparks et al.,
1995). Interestingly, Sparks et al. observed multi-
ple mitotic phosphorylation events occuring in simi-
lar timing of lamin B phosphorylation (Sparks et al.,
1995). Thus, our results show that phosphorylation
of SPTBN1 might negatively control the phosphory-
lation of NUMA1 acting in an antagonistic fashion
to the effect of LMNB2 phosphorylation. Similar to
NUMA1 the phosphorylation of NES is also posi-
tively related to the phosphorylation of LMNB2 and
negatively related to the phosphorylation of SPTBN1.
NES phosphorylation was shown to be an important
regulator for its organizationand dynamics during mi-
tosis in a neuronal progenitor cell line (Sahlgren et al.,
2001). Similar to NES the data reports LMNA phos-
phorylation to be lower in IL6 treated cells in NO,
but not in HO. Like LMNB2, LMNA phosphorylation
is associated with matrix disassembly in mitosis. Fi-
nally, phosphorylation of GTF2F1 is associated with
the stimulation of transcription (Solow et al., 2001).
The observed de-phosphorylation in IL6 treated cells
in NO therefore might correspond to a decreased tran-
scription rate.
Inference of Predictive Phospho-regulatory Networks from LC-MS/MS Phosphoproteomics Data
89

5 CONCLUSION
In this study, we used phosphoproteomics data de-
rived from LC-MS/MS to illustrate the inference of
phospho-regulatory networks based on the modeling
concept of the published TILAR algorithm. The ad-
vantage of this approach is that knowledge about reg-
ulating phosphatases and kinases serves as a knowl-
edge base to create a network structure template that
guides the inference. This way, prior-knowledge
can be automatically integrated to create biologi-
cally meaningful, intuitive network models represent-
ing the experimentally measured data. For the in-
ference of the PRN we used published data measur-
ing the changes in phosphorylation of proteins from
JAK2/STAT3 phosphorylation inhibitor WP1193 per-
turbed GSC11 cells treated with IL6 under NO and
HO conditions. In total, 21 DPPs were selected for
the inference with most of them related to the GO-
terms ’structural molecule activity’ and ’cell cycle’.
Our results suggest that the oxygen concentration has
an impact on IL6 induced changes in protein phos-
phorylation. While phosphorylation of most of the
selected DPPs does not change upon IL6 treatment in
NO, there is an increased phosphorylation in mitosis-
associated proteins such as LMNB2 and MARCKS in
HO. This exemplifies how PRNs can aid in the inter-
pretation of phosphoproteomics data. However, due
to the shortage of experimental data the derived hy-
potheses will have to be verified using additional ex-
perimental data.
ACKNOWLEDGEMENTS
This work was supported by the BMBF (Virtual Liver
Network, FKZ: 0315736) and the excellence gradu-
ate school Jena School for Microbial Communication
(JSMC).
REFERENCES
Dechat, T., Pfleghaar, K., Sengupta, K., Shimi, T., Shu-
maker, D. K., Solimando, L., and Goldman, R. D.
(2008). Nuclear lamins: major factors in the structural
organization and function of the nucleus and chro-
matin. Genes Dev, 22(7):832–853.
Efron, B., Hastie, T., Johnstone, I., Tibshirani, R., et al.
(2004). Least angle regression. The Annals of statis-
tics, 32(2):407–499.
Hecker, M., Goertsches, R. H., Engelmann, R., Thiesen, H.-
J., and Guthke, R. (2009a). Integrative modeling of
transcriptional regulation in response to antirheumatic
therapy. BMC Bioinformatics, 10:262.
Hecker, M., Lambeck, S., Toepfer, S., van Someren, E., and
Guthke, R. (2009b). Gene regulatory network infer-
ence: data integration in dynamic models-a review.
Biosystems, 96(1):86–103.
Hornbeck, P. V., Kornhauser, J. M., Tkachev, S., Zhang,
B., Skrzypek, E., Murray, B., Latham, V., and Sul-
livan, M. (2012). Phosphositeplus: a comprehen-
sive resource for investigating the structure and func-
tion of experimentally determined post-translational
modifications in man and mouse. Nucleic Acids Res,
40(Database issue):D261–D270.
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009).
Systematic and integrative analysis of large gene lists
using david bioinformatics resources. Nat Protoc,
4(1):44–57.
Jørgensen, C. and Linding, R. (2008). Directional and quan-
titative phosphorylation networks. Brief Funct Ge-
nomic Proteomic, 7(1):17–26.
Linding, R., Jensen, L. J., Pasculescu, A., Olhovsky, M.,
Colwill, K., Bork, P., Yaffe, M. B., and Pawson, T.
(2008). Networkin: a resource for exploring cel-
lular phosphorylation networks. Nucleic Acids Res,
36(Database issue):D695–D699.
Mallows, C. L. (1973). Some comments on c p. Techno-
metrics, 15(4):661–675.
Manno, S., Takakuwa, Y., Nagao, K., and Mohandas, N.
(1995). Modulation of erythrocyte membrane me-
chanical function by beta-spectrin phosphorylation
and dephosphorylation. J Biol Chem, 270(10):5659–
5665.
Nikitin, A., Egorov, S., Daraselia, N., and Mazo, I. (2003).
Pathway studiothe analysis and navigation of molecu-
lar networks. Bioinformatics, 19(16):2155–2157.
Nilsson, C. L., Dillon, R., Devakumar, A., Shi, S. D.-H.,
Greig, M., Rogers, J. C., Krastins, B., Rosenblatt,
M., Kilmer, G., Major, M., Kaboord, B. J., Sarra-
cino, D., Rezai, T., Prakash, A., Lopez, M., Ji, Y.,
Priebe, W., Lang, F. F., Colman, H., and Conrad, C. A.
(2010). Quantitative phosphoproteomic analysis of
the stat3/il-6/hif1alpha signaling network: an initial
study in gsc11 glioblastoma stem cells. J Proteome
Res, 9(1):430–443.
Pai, C. Y., Chen, H. K., Sheu, H. L., and Yeh, N. H. (1995).
Cell-cycle-dependent alterations of a highly phospho-
rylated nucleolar protein p130 are associated with nu-
cleologenesis. J Cell Sci, 108 ( Pt 5):1911–1920.
R Core Team (2014). R: A Language and Environment for
Statistical Computing. R Foundation for Statistical
Computing, Vienna, Austria.
Rombouts, K., Mello, T., Liotta, F., Galli, A., Caligiuri,
A., Annunziato, F., and Pinzani, M. (2012). Marcks
actin-binding capacity mediates actin filament assem-
bly during mitosis in human hepatic stellate cells. Am
J Physiol Cell Physiol, 303(4):C357–C367.
Sahlgren, C. M., Mikhailov, A., Hellman, J., Chou, Y. H.,
Lendahl, U., Goldman, R. D., and Eriksson, J. E.
(2001). Mitotic reorganization of the intermediate fila-
ment protein nestin involves phosphorylation by cdc2
kinase. J Biol Chem, 276(19):16456–16463.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
90

Seet, B. T., Dikic, I., Zhou, M.-M., and Pawson, T. (2006).
Reading protein modifications with interaction do-
mains. Nat Rev Mol Cell Biol, 7(7):473–483.
Smith, N. L. and Miskimins, W. K. (2011). Phosphorylation
at serine 482 affects stability of nf90 and its functional
role in mitosis. Cell Prolif, 44(2):147–155.
Solow, S., Salunek, M., Ryan, R., and Lieberman, P. M.
(2001). Taf(ii) 250 phosphorylates human transcrip-
tion factor iia on serine residues important for tbp
binding and transcription activity. J Biol Chem,
276(19):15886–15892.
Sparks, C. A., Fey, E. G., Vidair, C. A., and Doxsey, S. J.
(1995). Phosphorylation of numa occurs during nu-
clear breakdown and not mitotic spindle assembly. J
Cell Sci, 108 ( Pt 11):3389–3396.
Spiliotis, E. T., Kinoshita, M., and Nelson, W. J. (2005).
A mitotic septin scaffold required for mammalian
chromosome congression and segregation. Science,
307(5716):1781–1785.
Terfve, C. and Saez-Rodriguez, J. (2012). Modeling
signaling networks using high-throughput phospho-
proteomics. Adv Exp Med Biol, 736:19–57.
Xue, Y., Ren, J., Gao, X., Jin, C., Wen, L., and Yao, X.
(2008). Gps 2.0, a tool to predict kinase-specific phos-
phorylation sites in hierarchy. Mol Cell Proteomics,
7(9):1598–1608.
Yang, X.-J. (2005). Multisite protein modification and
intramolecular signaling. Oncogene, 24(10):1653–
1662.
Inference of Predictive Phospho-regulatory Networks from LC-MS/MS Phosphoproteomics Data
91
