
Automatic Feature Selection in the SOPFs Dissolution Profiles Prediction
Problem
J. E. Salazar-Jim´enez
1
, J. D. S´anchez-Carvajal
1
, B. Quiros-G´omez
2
and J. D. Arias-Londo˜no
3
1
I&S Research group, Faculty of Engineering, Universidad de Antioquia, Cll 70 No. 52-21, Medell´ın, Colombia
2
Unidad de Investigaci´on e Innovaci´on, Humax Pharmaceutical S.A., Calle 97B Sur No. 50 - 95,
La Estrella, Antioquia, Colombia
3
Dpt. Of Systems Engineering and Computer Science, Universidad de Antioquia, Cll 70 No. 52-21, Medell´ın, Colombia
Keywords:
Automatic Feature Selection, Bioequivalence, Drug Development, Drug Dissolution Profile Prediction, Solid
Oral Pharmaceutical Forms.
Abstract:
This work addressed the problem of dimensionality reduction in the drug dissolution profile prediction task.
The learning problem is assumed as a multi-output learning task, since dissolution profiles are recorded in non-
uniform sampling times, which avoid the use of basic function-on-scalar regression approaches. Ensemble-
based tree methods are used for prediction, and also for the selection of the most relevant features, because
they are able to deal with high dimensional feature spaces, when the number of training samples is small.
All the drugs considered corresponds to rapid release solid oral pharmaceutical forms. Six different feature
selection schemes were tested, including sequential feature selection and genetic algorithms, along with a
feature scoring procedure, which was proposed in order to get a consensus about the best subset of variables.
The performance was evaluated in terms of the similitude factor used in the drug industry for dissolution
profile comparison. The feature selection methods were able to reduce the dimensionality of the feature space
in 79.2%, without loss in the performance of the prediction system. The results confirm that in the dissolution
profile prediction problem, especially for different solid oral pharmaceutical forms, variables from different
components and phases of the drug development must be considered.
1 INTRODUCTION
The development of solid oral pharmaceutical forms
(SOPFs) must satisfy several requirements from tech-
nical, scientific and legal statements. Usually, the
whole development process is performed by adjust-
ing the design in the laboratory between the formu-
lation stage, and the verification of complying indus-
trial manufacturing scaling standards (Gibson, 2005)
(Moon, 2011). One of the more important technical
requirements for SOPFs, is to assure that the pro-
duct has an appropriate biopharmaceutical behavior
(BPB), which includes the estimated time of effect or
duration, among other important clinic pharmaceuti-
cal properties (Shargel et al., 2007). A key element
of the BPB evaluation is the reconstruction of a time
curve known as dissolution profile (DP), which pro-
vides the dissolution percentages of the drugs’ active
ingredient (AI) through time (Shargel et al., 2007).
In the generic pharmaceutical industry, the
development of SOPFs according to a Quality
by Design approach, must additionally satisfy a
pharmacokinetic-based measure known as Bioequi-
valence, which is a comparison between the DPs of
the generic drug and the reference product. The Bio-
equivalence lets generic laboratories to demonstrate
that their products are statistically similar in terms of
both, BPB and therapeutic properties, to those of the
reference drugs (Shargel et al., 2007). However, the
main drawback to get this objective, is the fact that the
DPs follow a no lineal behavior influenced by a large
number of variables including physical and chemi-
cal properties of the excipients and AIs, the interac-
tions that could occur between them, their respective
proportion in the formula, the manufacture param-
eters, the parameters of the dissolution test, among
others (Dokoumetzidis and Mahceras, 2006) (Ghayas
et al., 2013). Therefore, the task of obtaining a de-
sired DP sometimes becomes in the bottleneck of the
generic SOPFs design. Moreover, since the lack of
better techniques or optimization methods, the pro-
cess of getting a desired DP is approached by a “trial
52
Salazar JimÃl’nez J., Sà ˛anchez Carvajal J., Quiros-Gøsmez B. and Arias-LondoÃ
´
so J.
Automatic Feature Selection in the SOPFs Dissolution Profiles Prediction Problem.
DOI: 10.5220/0006141800520058
In Proceedings of the 10th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2017), pages 52-58
ISBN: 978-989-758-214-1
Copyright
c
2017 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

and error” guided formulation design, supported ex-
clusively by the experience, expertise and knowledge
of drug development scientists (Aguilar, 2013).
In the last fifteen years, many efforts have been
made in order to develop computational methods that
can provide a tool for the prediction/simulation of
DPs. Mechanistic and data-driven (phenomenologi-
cal) models have been used for this purpose (Siep-
mann and Siepmann, 2013) (Mendyk et al., 2015).
Mechanistic approaches are a more elegant and ac-
curate way to model the dynamic interaction among
the variables. However, they include many parame-
ters that are difficult to estimate and require a deep
understanding of every law governing the interac-
tion among all the variables involved in the disso-
lution process, and much of them are still unknown
(Aguilar, 2013). On the other hand, computational
intelligence methods, and more specifically machine
learning (ML) techniques, are able to generate mod-
els trough a data-driven paradigm, with the advan-
tage that no a priori knowledge about the interactions
among the variables is required (Ibri´c et al., 2012).
Most of the ML-based approaches for DP prediction
focus their analysis in the use of Artificial Neural Net-
works (ANN) with different topologies. e.g., in (Shao
et al., 2007) a comparison between neurofuzzy logic
and a basic ID3 decision tree approaches is presented.
A total of 14 variables were included (4 formulation
variables, 2 process variables, and 8 tablet proper-
ties). No information about the number of AIs in-
cluded in the experiments is given by the authors. The
paper concluded that both models are able to provide
useful knowledge about the cause-effect relationships
among the variables and the quality of the product.
In another case study (Ibri´c et al., 2012), a review
of the application of ANNs in the formulation and
evaluation of modified release dosage forms is pre-
sented. Multi-layer perceptron and Elman neural net-
works are the most employed methods according to
the revision. In all the cases cited, the models are used
to predict DPs in highly controlled environments, i.e.
the data contain only one AI and few design varia-
bles, specially features related to formula composi-
tion. A more recent approach presented in (Mendyk
et al., 2015), compares the performance of ANN and
Genetic Programming (GP) in the modeling of drug
dissolution from the dosage form. The data set con-
tained results of dissolution tests carried out for 5 var-
ious formulations of lipid extrudates. Only two varia-
bles were included in the analysis. The authors found
GP to be the most robust model for DP prediction.
Bearing this in mind, the main limitation of the
computer-aided dissolution profiles prediction sys-
tems, to become in useful tools that really support the
design of a wider kind of SOPFs, is that they often
focus on specific dissolution phenomena, including
very scarce features and trained using data from only
one or maximum three different AIs. Therefore, they
serve limited purpose and their use in real develop-
ment environments could be considered narrow.
Building a ML-based tool able to simulate DPs of
different SOPFs, requires to consider a larger num-
ber of variables involved in the dissolution process,
because the dynamic response (DP) of some drugs
can drastically change by variations in features that
does not affect other kind of drugs. Nevertheless, by
increasing the number of variables to be considered
by complex data-driven models (such as ANN, whose
parameters increase exponentially with respect to the
number of variables), the model trained can likely be
affected by the curse of dimensionality, and overfits
to the training data.
From an statistical point of view, the prediction
of a DP from a set of formulation variables, corre-
sponds to a functional regression problem known as
Function-on-Scalar Regression (FoSR) (Reiss et al.,
2010), i.e., a regression problem where the respon-
ses are functions and the predictors are scalars. In
order to overcome the curse of dimensionality in the
DP prediction problem, a method for dimensionality
reduction on FoSR is required; however this is an al-
most unexplored field in the state of the art, especially
for cases where the sampling times are not uniform
among the samples, which is precisely the case of
DPs, since the sampling times typically used in the
dissolution tests, are not uniform trough time and de-
pend on the duration or desired effect of the specific
drug being designed.
Bearing this in mind, an alternative way to ad-
dress the DP prediction problem, is to use a multi-
output ML-based approach, where the different va-
riables involved in the drug design and test, along
with the target dissolution times are used as inputs,
and the percentages of dissolution for the same tar-
get times are considered as outputs. This alternative
is suggested in (Contia and O’Hagan, 2010) for com-
plex non-uniform sampled dynamic models. Such a
model can be used as a wrapper criterion for feature
selection techniques, in order to reduce the number
of variables analyzed, avoid the overfitting of the pre-
diction model, and select the most relevant features
for the DP prediction problem.
In this sense, the present work explores the use
of heuristic-based methods, in order to address the
dimensionality reduction in the SOPFs’ DP predic-
tion problem. All the drugs considered correspond
to rapid release SOPFs, which have similar pharma-
cokinetics and are the most frequently type of SOPFs
Automatic Feature Selection in the SOPFs Dissolution Profiles Prediction Problem
53

produced by the pharmaceutical industry, specially
by generic laboratories (Shargel et al., 2007) (Qiu
and Zhou, 2011). The feature selection process eval-
uates 6 methods based on sequential feature selec-
tion and genetic algorithms, coupled with two super-
vised techniques, multi-output bagging of trees (BT)
and extremely randomized trees (ERT). Additionally,
BT and ERT are used themselves as feature selection
technique. In previous experiments, ANN and multi-
output support vector regressors were also evaluated,
but tree-based ensemble methods achieved better per-
formance. In order to follow a multi-output approach,
all the DPs were restricted to have the same number
of sampling times.
The rest of the paper is organized as follows: sec-
tion 2 presents the set of variables included, the re-
gression models and the feature selection techniques
employed. Section 3, describe the dataset and the va-
lidation methodology. Section 4 presents the results
obtained and finally section 5 includes some conclu-
sions.
2 METHODS
2.1 Variables Definition
A set of 168 variables were included for the study.
They were added for reasons of theoretical relevance
and availability. The input variables were classified
into 8 different groups, each one considered to be
essential in SOPFs’ DPs analytics:
1. Galenic features (number of initial features = 88):
Features Associated with drug design and formula
composition (e.g. %AI, and one hot encodingvec-
tor codifying the presence of a specific excipient,
etc.)
2. Pharmacotecnic features (number of initial fea-
tures = 11): Corresponding to variables, condi-
tions and parameters related to the manufacturing
process and drug development equipment (e.g.
mixing time, humidity, tablet hardness, etc.)
3. Final form physical features (number of initial
features = 16): Variables associated to dimen-
sional measures of the final SOPF (e.g. tablet
thickness, length, etc.)
4. AIs’ physicochemical features (number of initial
features = 11): set of features associated with the
AIs’ physical and chemical characteristics (e.g.
AI molecular weight, logP, rotable bonds, etc.)
5. AIs’ pharmaco-molecularfeatures (number of ini-
tial features = 10): Variables related to the pre-
sence of specific molecular/chemical groups of
pharmaceutical interest (number of AI hydroxyls,
number of AI amines, etc.)
6. Formula physicochemicalfeatures (numberof ini-
tial features = 13): Similar to group 5, some
physicochemical characteristic of the tablet func-
tional components, (e.g. binder solubility in wa-
ter, surfactant particle size, etc.)
7. Drug dissolution test related features (number of
initial features = 16): All parameters and features
related to the analytic measure of %AI (e.g. sol-
vent used, analytic method, rotational speed, etc.).
8. Sampling times features (number of initial fea-
tures = 3): Corresponding to the 3 tested times
during the reconstruction of the PD.
The output or dependent variables correspond to
the dissolution percentage for every sampling time.
2.2 Prediction Models
Two different classification models were used in this
work for DP prediction. In first place a multi-output
bagging of Trees (BT) was used as predictor. This
corresponds to a set of decision trees estimated on
bootstrap samples extracted from the training data.
Ensemble methods have demonstrated to reach
comparable performance to complex parametric and
kernel based methods. Moreover, in this study they
were selected after a set of experiments where the
performance of BT was compared to ANN and multi-
output support vector regressors. For the dataset used
in this work, ensemble-based tree methods provided
the best results. This behavior could be explained be-
cause ANN and SVR require a larger number of sam-
ples to get a successful training phase, especially in
high dimensional feature spaces.
Additionally, a more computational efficient tree-
based ensemble method was also used. Extremely
Randomized Trees (ERT) (Geurts et al., 2006) are a
class of tree-based ensemble methods, where for each
decision node, a random subset of candidate features
is used (instead of the whole set), and thresholds are
drawn also at random for each candidate feature. The
best of these randomly-generatedthresholds is picked
as the splitting rule. ERT provides a similar perfor-
mance than Random Forest, but they are computa-
tionally more efficient (Geurts et al., 2006), which is
a desirable property especially when the methods are
going to be used as criterion for feature selection al-
gorithms.
In all the cases, the models were fed with the vari-
ables involved in the drug design and test, along with
the target dissolution times, and they were asked to
produce the corresponding dissolution percentage per
BIOINFORMATICS 2017 - 8th International Conference on Bioinformatics Models, Methods and Algorithms
54

every of the input times. Since the learning strategy
corresponds to a multi-ouput learning paradigm, all
the samples were restricted to have a constant num-
ber of times. As it as pointed before, in this work the
number of times was fixed to 3, since according to the
methodology for dissolution test (FDA, 1997), a DP
must be evaluated with minimum 3 sampling times.
2.3 Feature Selection
Two Feature Selection Techniques (FSTs) were eval-
uated using wrapper methodsas selection criteria. Se-
quential Forward Selection (SFS) and Genetic Algo-
rithm (GA), were used as search algorithms.
• SFS is a bottom-up selection method which build
up a set of p features incrementally, starting with
the empty set and adding new features to the fea-
ture set one at a time, until the final set is reached
(Webb, 2003). Suppose that during the iteration
t, p
1
features have been included into the selected
set of features X . For each of the features ν
j
not
yet selected, the criterion function J
j
= J(X + ν
j
)
is evaluated. The feature that yields the maximum
value of J
j
is chosen as the one that is added to
the set X . When the best improvement makes the
feature set worse, or when the maximum allow-
able number of features is reached, the algorithm
terminates (Webb, 2003).
• GAs are heuristic optimization and search tech-
niques inspired by principles of genetics and the
natural selection process. A GA allows a pop-
ulation composed of many individuals to evolve
under specific selection rules to a state that maxi-
mizes the “fitness” (criterion function) (Haupt and
Haupt, 2004). In this work, a binary GA was
used, where each individual (a candidate subset of
features), was represented as a vector of binary-
valued components of length equal to the total
number of features. A ’1’ value in the j-th posi-
tion of one individual means that the j-th feature
is included in the candidate subset represented by
such an individual, otherwise such features is not
included. After convergence, the GA provides a
solution for the optimization problem, which in
this case corresponds to the final subset of se-
lected features.
The criterion used for SFS and GA corresponds to
a wrapper method based on the two ensemble-based
trees learning algorithms described before (multi-
output ERT and BT). Additionally, ERT and BT were
used themselves as feature selection methods. This
type of models can be used as feature selectors, since
the learningstrategy in which they are based on, select
one feature to split the feature space in every internal
node of the tree. Therefore, the number of times that
a specific feature is used as splitting criteria (or sim-
ply to determine whether it is used for any internal
node or not), is directly proportional with the rele-
vance of such a feature for the prediction problem.
In this scenery, BT has and advantage over ERT, be-
cause BT analyses all the features in every internal
node, whilst ERT uses a random subset of candidate
features per node. However, the performance of both
models as feature selectors was evaluated. In total six
different FST methods were implemented. In all the
cases the main criterion for selection was the average
of the similitude factor f
2
given by
f2 = 50log
100
1+
1
n
n
∑
t=1
(R
t
− T
t
)
2
!
−0.5
(1)
where R
t
is the percentage of AI dissolved from a ref-
erence drug at a time t, and T
t
is the predicted value
by the model at the same time. n is the number of
sampling times. For all the samples in this work,
n = 3. Similitude factor f
2
was used, since it is the
criterion suggested by the US Food and Drug Admin-
istration (FDA) for dissolution test (FDA, 1997). A
detailed analysis of the f
2
factor, can show that it cor-
responds to a nonlinear mapping of the mean square
error, which is one of the classical measures for the
evaluation of regression models. f2 ranges in the in-
terval (−∞, 100], being 100 a perfect match between
the compared DPs.
The set of selected features must corresponds to
the one that provides the best average f
2
. Taking into
account that every FST would provide slightly dif-
ferent subsets of features, and in order to get a con-
sensus, a ranking strategy was implemented. Differ-
ent subsets of candidate features were finally tested,
according to their ranking, aiming to determine the
performance obtained by each one and the percentage
of reduction that the methods were able to find. The
ranking strategy consisted in assigning one score to
every feature, according to the number of times that
each one was selected during the several runs of the
simulations. Moreover, the score also assign higher
values to features that were selected into small sub-
sets. The aim of this strategy is to find smaller subsets
of features with the highest accuracies. Taking into
account that every FST was evaluated multiple times,
according to the validation methodology (see section
3.2), the first step was to estimate a relevance factor
for each feature, which corresponds to the percentage
of times that the feature was included in the selected
subset, taking into account the total number of fea-
tures that were selected in any of the repetitions. For-
Automatic Feature Selection in the SOPFs Dissolution Profiles Prediction Problem
55

mally, let g
ef
∈ {0, 1}, an indicator variable that takes
the value of 1, if the input variable f was included in
the final subset during the experiment e, and 0 other-
wise. The score of the feature f is given by
S
f
=
∑
n
e
e=1
g
ef
∑
n
e
e=1
∑
n
d
j=1
g
ej
(2)
where n
e
is the number of experiments or repetitions
during the validation, and n
d
the number of varia-
bles. Additionally, the FST were evaluated for dif-
ferent number of trees in the bagging, therefore the
final score per feature was estimates as the average of
the scores obtained for the different number of trees.
Several subsets of features were finally tested by
changing the minimum allowed score to be conside-
red a relevant feature.
3 EXPERIMENTAL SETUP
3.1 Data Set
Data was provided by Humax Pharmaceutical S.A.
and consisted of 658 records from about 60 different
products assays and about 50 AIs. Each formulation
assay or “record” had an associated output PD com-
posed by at least three sampling times. All the record-
ings where standardized to three sampling times, en-
suring that only one of the measurement be after 85%
dissolution (as suggested by FDA (FDA, 1997)). As it
was pointed out, all the drugs considered correspond
to rapid release SOPFs.
3.2 Validation Methodology
All the experiments were performed using a boot-
strapping validation strategy, with 70% of the sam-
ples for training and 30% for validation. Ten repeti-
tions for every experiment were carried out. The opti-
mal number of trees was selected according to a grid
search ranging from 10 to 100. In order to increase the
reliability of the subset of features finally selected, the
feature selection strategies were evaluated with differ-
ent number of trees, and all their results were taken
into account, as it was explained in section 2.3.
The performance of the methods were evaluated
according to the similitude factor f
2
(1). According
to the Guidance for Dissolution Testing of Immediate
Release SOPFs provided by the FDA (FDA, 1997),
two DPs are considered similar if the f
2
factor be-
tween them is greater than 50. Therefore, an addi-
tional error measure was estimated, by averaging the
number of times that the predicted DP did not exceed
such threshold. This error measure was called Err
f
2
4 RESULTS AND DISCUSSION
Table 1 shows the performance obtained using the
whole set of features described in section 2.1, using
an ERT-based predictor. This results are going to be
considered the base line for comparison purposes.
Table 1: Best results obtained for the whole set of features
using ERT.
Number of trees Average f
2
Average Err
f
2
10 58.93± 14.80* 28.03%± 3.8
20 60.20± 14.82 25.25%± 1.6
30 59.28± 14.87 27.17%± 2.9
40 59.04± 14.91 27.58%± 3.0
50 58.88± 15.23 29.14%± 3.4
60 59.26± 15.18 28.08%± 3.2
70 59.76± 14.82 26.41%± 2.0
80 59.74± 14.93 26.92%± 1.8
90 58.98± 14.79 27.73%± 2.1
100 59.27 ± 14.85 26.62%± 1.9
*mean ± standard deviation.
From table 1, is possible to observe that the av-
erage f
2
is not very sensitive to the number of trees.
However, for the largest number of trees evaluated,
the results are more stable in both, f
2
and Err
f
2
.
Therefore, the experiments for features selection were
performed using 80, 100 and 120 trees for every
method.
Figures 1 and 2 shows the percentage of reduc-
tion and similitude factors obtained for the different
FST evaluated, and changing the score threshold for
the variable inclusion. As a higher threshold is cho-
sen, more features are excluded, because the criterion
becomes stricter. This analysis allows to show how
different the subsets of features are per every run of
the different FSTs evaluated. If in every run of a FST
the subset of features is almost the same, such FST
is quite consistent and is able to select the best subset
of features even with a low score threshold. This is
the case of FSF-based selectors. Otherwise, if a FST
produce different results in every run, a larger score
threshold for the variable inclusion is required in or-
der to get a consensus.
The figures 1 and 2 shows the results before all the
methods started to reduce their performance, because
most of the features, or all of them, were excluded.
From figure 1 is possible to observe how SFS-based
methods are able to identify the “best” subset fea-
tures, even for small scoring threshold. This means
that the non-relevant features are excluded in almost
every run of the algorithm. Additionally, when BT
was used as wrapper criterion, the performance of the
selected subset of feature reach a higher f
2
(see fig-
BIOINFORMATICS 2017 - 8th International Conference on Bioinformatics Models, Methods and Algorithms
56
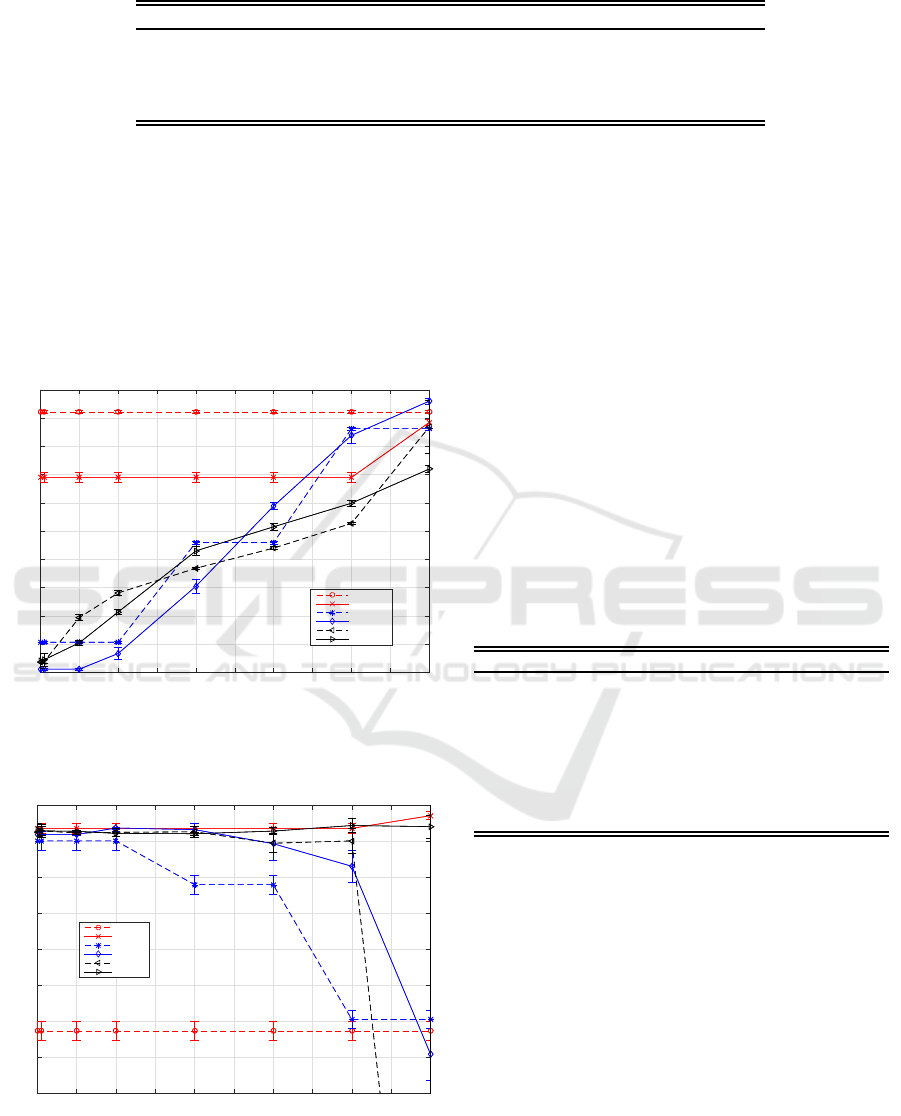
Table 2: Best results after the feature selection stage.
FST Average f
2
Average Err
f
2
Percentage of reduction
TB-120 60.03± 15.31 25.95%± 2.9 79.2%
SFS-TB-80 59.73± 15.00 26.29%± 2.4 89.2%
SFS-TB-120 59.84 ± 15.03 26.42%± 2.9 86.9%
TB-100 59.72± 15.07 26.67%± 2.3 70.8%
ure 2). Besides when BT was used alone as feature
selector, the performance was also high. This behav-
ior could be explained by the fact that BT evaluates all
the features in every decision node, whilst ERT evalu-
ates only a subset of randomly selected features. This
randomly selection provides ERT with a better com-
putational efficiency, but reduces their performanceas
feature selector.
Score
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
% of Features Reduced
0
10
20
30
40
50
60
70
80
90
100
Reduction Profile
SFSERT
SFSTB
GAERT
GATB
ERT
TB
Figure 1: Percentage of reduction obtained for each FST
evaluated and different thresholds in the score.
Score
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
mean f2
52
53
54
55
56
57
58
59
60
Performance Profile
SFSERT
SFSTB
GAERT
GATB
ERT
TB
Figure 2: Average similitude factor obtained for each FST
evaluated and different thresholds in the score.
Table 2 shows the best results obtained using the
different FSTs, and scoring thresholds. It is impor-
tant to note how feature selection using TB was able
to achieve a similar performance than those in table 1,
but with a reduction of 79.2% in the number of vari-
ables. Moreover, other SFS-based methods were also
able to yield similar performances, although some au-
thors assert that this kind of methods are suitable for
classification but not for regression problems (Caste-
llano and Fanelli, 2000). . On the other hand, the use
of GA as search method did not produce satisfactory
results. Furthermore, its computational cost makes it
a less interesting option as feature selector.
Finally, table 3 shows how the feature dimensi-
onality reduction is split among the different groups
of variables considered. This is an interesting result,
since it confirms that almost all the groups conside-
red are relevant for the prediction of DPs, whenever
the multi-output regression system have to work with
different SOPFs.
Table 3: Percentage of reduction according to every group
of variables analyzed.
Group of variables Percentage of reduction
Galenic 87.5%
Pharmacotecnic 45.5%
Final form 81.3%
AIs’ physicochemical 90.9%
AIs’ pharmaco-molecular 100%
Formula 76.9%
Dissolution test 81.3%
Sampling times 0.0%
5 CONCLUSIONS
The DP prediction problem is a complex task that can
be understood as a functional regression where the
responses are functions and the predictors are scalars.
In the case of DPs, the sampling times used dur-
ing dissolution test vary significantly from one drug
to another, introducing additional challenges to the
modeling process. From a machine learning perspec-
tive, this problem can be addressed as a multi-output
learning task. The results show that the ensemble-
tree based methods are able to provide DP predictions
that, in average, exceed the minimum allowed value
to consider two DPs as similar (according to the US
Food and Drug Administration).
Automatic Feature Selection in the SOPFs Dissolution Profiles Prediction Problem
57

DP prediction from datasets that include many dif-
ferent solid oral pharmaceutical forms, requires the
introductionof features from different componentand
phases of the drug development process. This fact in-
creases the dimensionality of the feature space used
by the learning algorithm, whichcan be problematic if
the size of the training set is small. In this context, the
use of sequential feature selection techniques coupled
to wrapper criteria based on multi-output ensemble-
tree methods, becomes in an interesting alternative
that allows the identification of relevant and reliable
subsets of features.
The results showed that a significantly reduced
feature set can be found, and also that such subset is
able to provide a similar performance than the com-
plete set of features. Moreover, taking into account
that the database used contains different SOPFs, the
automatic selection also showed that almost all the
groups of variables considered, were found to be rel-
evant for the prediction of DPs.
Additional work must be done to include other
kind of ML based feature selection methods that can
also be adapted to this problem, as well as multi-input
multi-output feature extraction techniques.
ACKNOWLEDGEMENTS
This research was supported by Project No. CODIIA
15-1-01 funded by Universidad de Antioquia,
Medell´ın, Colombia and Humax Pharmaceutical S.A.
The authors had full access to all of the data in this
study and take complete responsibility for the in-
tegrity of the data and the accuracy of the data anal-
ysis, certifying that all financial and material support
for the conduct of this study and/or preparation of this
manuscript is in compliance with ethical standards.
Moreover, the authors declare having taken part in the
research presented in this paper, having reviewed and
approved the manuscript being submitted. The au-
thors would like to thank Alba Ceballos and Rodrigo
Ochoa for useful discussions.
REFERENCES
Aguilar, J. (2013). Formulation tools for pharmaceutical
development, volume 44. Woodhead Publishing.
Castellano, G. and Fanelli, A. (2000). Variable selection
using neural-network models. Neurocomputing, 31(1-
4):1–13.
Contia, S. and O’Hagan, A. (2010). Bayesian emulation
of complex multi-output and dynamic computer mod-
els. Journal of Statistical Planning and Inference,
140(3):640–651.
Dokoumetzidis, A. and Mahceras, P. (2006). A century
of dissolution research: from noyes and whitney to
the biopharmaceutics classification system. Int. J.
Pharm., 321(1-2):1–11.
FDA, U. (1997). Guidance for industry: Dissolution testing
of immediate-release solid oral dosage forms. Food
and Drug Administration, Center for Drug Evaluation
and Research (CDER).
Geurts, P., Ernst, D., and Wehenkel, L. (2006). Extremely
randomized trees. Machine Learning, 63(1):3–42.
Ghayas, S., Sheraz, M., Anjum, F., and Baig, M. (2013).
Factors influencing the dissolution testing of drugs.
Pak. J. Heal. Res., 1(1):1–11.
Gibson, M. (2005). Technlogy Transfer: An international
good practice guide for pharmaceuticals and allied
industries. DHI Publishing LLC.
Haupt, R. L. and Haupt, S. E. (2004). Practical genetic
algorithms. John Wiley & Sons.
Ibri´c, S., Djuriˇs, J., Parojˇci´c, J., and Djuri´c, Z. (2012). Ar-
tificial neural networks in evaluation and optimization
of modified release solid dosage forms. Pharmaceu-
tics, 4:531–550.
Mendyk, A., Gres, S., Jachowicz, R., Szlk, J., Polak, S.,
Winiowska, B., and Kleinebudde, P. (2015). From
heuristic to mathematical modeling of drugs dissolu-
tion profiles: Application of artificial neural networks
and genetic programming. Comput. Math. Methods
Med., 2015:1–9.
Moon, S. (2011). Pharmaceutical Production and Related
Technology Transfer. World Health Organization.
Qiu, Y. and Zhou, D. (2011). Understanding design and
development of modified release solid oral dosage
forms. J. Valid. Technol., 17(2):2332.
Reiss, P., Huang, L., and Mennes, M. (2010). Fast function-
on-scalar regression with penalized basis expansions.
The International Journal of Biostatistics, 6(1):Article
28.
Shao, Q., Rowe, R., and York, P. (2007). Comparison
of neurofuzzy logic and decision trees in discover-
ing knowledge from experimental data of an imme-
diate release tablet formulation. European Journal
of Pharmaceutical Sciences: Official Journal of the
European Federation for Pharmaceutical Sciences,
31(2):129136.
Shargel, L., Wu-pong, S., and Yu, A. (2007). Applied Bio-
pharmaceutics & Pharmacokinetics. McGraw-Hill’s
ACCESPHARMACY, 5th edition.
Siepmann, J. and Siepmann, F. (2013). Mathematical mod-
eling of drug dissolution. Int. J. Pharm., 453(1):1224.
Webb, A. R. (2003). Statistical pattern recognition. John
Wiley & Sons, 2nd edition.
BIOINFORMATICS 2017 - 8th International Conference on Bioinformatics Models, Methods and Algorithms
58
