
Comparative Study on Data Mining Techniques Applied to Breast
Cancer Gene Expression Profiles
Sérgio Mosquim Júnior
1,2
and Juliana de Oliveira
1
1
School of Sciences, Humanities and Languages, São Paulo State University, Av. Dom Antonio, 2100, Assis - SP, Brazil
2
Uppsala University, Uppsala, Sweden
Keywords: Data Mining, Breast Cancer, Decision Trees, Artificial Neural Networks.
Abstract: Breast cancer has the second highest incidence among all cancer types and is the fifth cause of cancer
related death among women. In Brazil, breast cancer mortality rates have been rising. Cancer classification
is intricate, mainly when differentiating subtypes. In this context, data mining becomes a fundamental tool
to analyze genotypic data, improving diagnostics, treatment and patient care. As the data dimensionality is
problematic, methods to reduce it must be applied. Hence, the present study aims at the analysis of two data
mining methods (i.e., decision trees and artificial neural networks). Weka® and MATLAB® were used to
implement these two methodologies. Decision trees appointed important genes for the classification.
Optimal artificial neural network architecture consists of two layers, one with 99 neurons and the other with
5. Both data mining techniques were able to classify data with high accuracy.
1 INTRODUCTION
According to the Brazilian National Cancer Institute
(INCA), cancer is the name given to more than 100
diseases that have a disorderly growth of cells
(malign) that penetrate tissues and organs, being
able to spread to other regions of the body in a
process called metastasis (Brasil, 2016).
According to estimates from the World Health
Organization (WHO) from 2011, cancer now is
responsible for more deaths than any other coronary
diseases. The constant changes in global
demographics and epidemiology lead to an increase
of the disease in the next decades, estimating 20
million new cases annually in 2025. In 2012, it was
estimated that 14.1 million new cases and 8.2
million deaths occurred globally (Ferlay et al.,
2015).
Breast cancer is the type of cancer which
presents the second highest incidence in the world
and is the most frequent in women, with 1.67
million new cases diagnosed in 2012. This value
corresponds to 25% of the total cancer diagnoses in
that year. Breast cancer still is the fifth cause of
death among several cancer types (Ferlay et al.,
2015).
In an analysis made by Desantis et al., (2015)
with data from GLOBOCAN, i.e. a project
developed by the International Agency for Research
on Cancer whose objective is to provide recent
incidence, mortality and prevalence estimates for the
most important types of cancer, it is discussed that
the highest incidences of breast cancer around the
world continue to be observed in the less developed
countries. It is estimated that nearly 1.7 million new
cases and 521,900 deaths were attributed to breast
cancer in the world in 2012.
Both in developed countries and in low to
medium income countries there has been an increase
in breast cancer incidence. This may be a result of
several factors, like changes in eating habits,
increase in obesity, hormone replacement therapy,
physical inactivity, among others (DeSantis et al.,
2015).
Despite this increase in breast cancer incidence,
mortality rates have decreased with time, especially
in developed countries. This tendency is a result of
more frequent mammographs and improvements in
treatment for example (DeSantis et al., 2015).
However, in countries like Brazil, Colombia,
Ecuador, Egypt, Guatemala, Japan, Kuwait, Mexico
and others, mortality rates increases, which reflects
and increase in incidence and, in some cases, the
limited access to treatment (DeSantis et al., 2015).
According to INCA, 57,960 new cases of breast
cancer are expected in 2016, with an estimate risk of
168
Mosquim JÞnior S. and de Oliveira J.
Comparative Study on Data Mining Techniques Applied to Breast Cancer Gene Expression Profiles.
DOI: 10.5220/0006170201680175
In Proceedings of the 10th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2017), pages 168-175
ISBN: 978-989-758-214-1
Copyright
c
2017 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

56.20 cases in every 100 thousand women. Without
taking into account non melanoma skin tumors,
breast cancer ranks first and most frequent in the
South, Southeast, Northeast and Center of Brazil.
Cancer classification has been a central topic in
treatment research. The most classical approach is
based on tumor morphology, which presents
limitations such as a strongly biased identification
by specialists and also difficulties in distinguishing
between subtypes (Liu et al., 2004). In this context
data mining appears as a means of facilitating
decision making processes by health professionals
when it comes to diagnosis, treatment and patient
care (Tseng et al., 2015).
Data mining belongs to a stage in the Knowledge
Discovery in Database (KDD) process (Tseng et al.,
2015). It is defined as the process of discovering
hidden patterns in data, which can take place
automatic or semi automatically (Witten et al.,
2011).
The vast applications of cDNA and
oligonucleotide microarrays, with complete genomic
expression, scanning more than 40,000 clones in a
single experiment, made possible the development
of a new era of molecular genomics. At the same
time, they’re generating vast amounts of data.
Molecular expression based classification continues
to be a challenge partly due to the different
microarray platforms, identification methods,
scanners, image analysis tools but also currently
available classification algorithms. Moreover,
there’s a growing number of algorithms that are
being developed for analysis of high quality
microarray data (Greer and Khan, 2004).
Due to the high cost, genetic data are usually
collected from a limited number of patients.
Therefore, there’s a need for choosing the most
relevant information among the available data.
Irrelevant gene removal can contribute to the
reduction of noise, confusion and complexity.
Besides, it increases the chances of identifying
important genes, classifying diseases and predicting
several outcomes (e.g., type of cancer). Several
computacional strategies have been applied to gene
expression classification problems (Shah and
Kusiak, 2007).
Learning methods constitute an automatic and
intelligent technique, which has been used widely
for solving different real and complex situations.
Since its introduction in bioinformatics, learning
approaches have helped to speed up diverse
researches. Since they are inexpensive and efficient,
its applications have become more popular and
constantly growing (Liu et al., 2004).
Usually, there are two different learning
schemes, supervised and unsupervised learning. In
the first one, the output is given, or there is some
type of previous knowledge about the data. On the
second one, however, there is no previous
knowledge about the data. General tasks performed
are classification, characterization and clustering.
The supervised approach is the most used in
biological problems where two sets of samples are
presented. The program, then, must generate a
classifier which is able to distinguish between these
two datasets. Then, it can be used as a base for the
classification of unseen data (Liu et al., 2004).
In an article published by Ahmad et al. (2015), it
was mentioned that when analyzing data mining in
healthcare, classification is one of the most popular
methods. The authors then follow by giving a list of
the most commonly used classification algorithms in
healthcare, such as K Nearest Neighbor (KNN),
Decision Trees, Support Vector Machines, Artificial
Neural Networks and Bayesian Methods (Ahmad et
al., 2015).
Decision Trees (DT) are considered to be one of
the most popular approaches when it comes to
classifiers. They can be built from data which is
already available in several fields. Every non leaf
node denotes a test to be performed, while branches
are outcomes. The tree ends in a leaf node, which
represents a class label. The most common use of
DTs is to calculate conditional probabilities. They
allow for class separation based on information gain.
The main advantages presented by this method are
the fact that DTs are self-explanatory, easy to
follow, the ability to handle nominal and numeric
attributes, ability to handle missing values.
However, they also present several disadvantages.
Most algorithms require the trees to have discrete
values, for they use the divide and conquer method.
Their performance gets lower the more complex is
the interaction among attributes. In that way, other
classifiers can describe the relationship among
variables in a way DTs would make it really
challenging (Ahmad et al., 2015).
Artificial Neural Networks (ANN), on the other
hand, were considered to be the best classification
algorithm before the introduction of methods such as
DTs and Support Vector Machines. This allowed
them to be one of the most widely used in several
different fields. They have widely used in supporting
diagnosis of diseases such as cancers and in
predicting outcomes. Their basic elements are
neurons (also called nodes), which are
interconnected and work in parallel to produce
outcome functions. The main ability behind ANNs is
Comparative Study on Data Mining Techniques Applied to Breast Cancer Gene Expression Profiles
169

that they are able to minimize error by adjusting the
connection weights and by making changes in its
structure. One of their main advantages is that they
can properly handle noisy data and classify data
different from the one used for training. However,
they require many parameters, including the number
of hidden layer nodes (empirically determined), the
learning process is computationally intense and time
consuming, and they do not provide any details
about the phenomenon being investigated.
Especially when it comes to the determination of
parameters, the performance is entirely dependent on
these factors (Ahmad et al., 2015).
The process of knowledge discovery can be
defined in five different steps, i.e., selection,
preprocessing, transformation, data mining and
interpretation. The preprocessing stage refers to the
removal of information which is not necessary for
the process and also cleans the data (Ahmad et al.,
2015). Shah and Kusiak (2007) mentioned that the
removal of irrelevant genes may contribute to noise
and complexity reduction while it also increases the
chances of identifying relevant genes. In most cases,
before using any data mining technique, a
dimensionality reduction approach is applied. It aims
at improving the performance of the method,
preventing overtraining and data is more easily
comprehended (Aguiar-Pulido et al., 2013).
On the above, the present work aims to compare
different data mining techniques (i.e., DT and ANN)
with respect to gathering high quality information on
breast cancer gene expression data.
2 METHODOLOGY
The data used in the present study consist of
publicly available breast cancer gene expression
data. Considering the KDD process described above,
a preprocessing and cleaning stages had to take
place before data mining itself. This consisted of
compiling the data, removing empty cells and using
a couple of filters in order to reduce dimensionality.
These filters were based on variance and entropy.
After this stage, the two different methods (DTs and
ANNs) were applied to the data and the results
analyzed and compared.
2.1 Data Acquisition
The invasive breast cancer gene expression (BRCA)
data were obtained from The Cancer Genome Atlas
(TCGA), which aims at accelerating the
comprehension of the molecular bases behind cancer
through the application of genomic analysis
techniques. The TCGA is a collaboration between
The National Cancer Institute and The Nacional
Human Genome Research Institute (EUA, 2016).
The present data corresponds to level 3 gene
expression data (i.e., expression values). These
values have already been normalized through
Lowess methodology when the data were acquired.
Typically, the first transformation applied to
expression data adjusts the individual hybridization
intensities to balance them, so meaningful biological
comparisons can be made. In addition to this
normalization, the expression rations must also be
normalized using Log2 values.
2.2 Preprocessing
The downloaded data were presented in text files,
one for each patient. These genes and their
expression values in these files were read and copied
into a spreadsheet by using a program written in
MATLAB®. This spreadsheet was then subjected to
different filters to remove irrelevant genes. The first
one detected empty values and removed the
corresponding genes, so there would be no errors
when reading the table. A second set of filters aimed
at removing genes with low variance and low
entropy values, which corresponds to noise in the
data.
2.3 Data Mining
The above mentioned data mining techniques (i.e.
DTs and ANNs) were performed on two different
pieces of software, Weka® and MATLAB®. The
DT were performed on both, while ANN was only
performed on MATLAB®.
2.3.1 Decision Trees
Oncologists classify different tumors based on
biopsy and other criteria. DT predict the class to
which a certain instance belongs. It is a simple
classifier, which is an advantage to this method
(Kingsford and Salzberg, 2008).
These trees are built splitting the data in two
parts, a training set that is used for the induction of
the tree, and a test set, which is used to check the
precision of the provided solution. After induction
the trees are used to classify unseen samples. It is a
hierarchical structure consisted of nodes (root,
internal node and terminal node) and directed lines.
Precise predictions can be achieved given adequate
training (Podgorelec et al., 2002; Kingsford and
BIOINFORMATICS 2017 - 8th International Conference on Bioinformatics Models, Methods and Algorithms
170

Salzberg, 2008; Aguiar-Pulido et al., 2013). The
classification task is quite simple. Starting at the
root, the testing condition is applied on the instances
and they follow the branches according to the
results. This will lead to an internal node, where the
same procedure is repeated, or to a leaf, where class
attribution is defined (Aguiar-Pulido et al., 2013).
On Weka® there are different tree algorithms. In
these case, two algorithms performed better, J48 and
REPTree. J48 uses the concept of entropy with a
training set, and every feature is used to make a
decision, which takes place by splitting the data into
smaller data sets. It is a pruned or unpruned tree
derived from the C4.5 tree. It uses information gain
to determine how much a property can separate the
training data according to the classification (Hall et
al., 2009 and Sa'di et al., 2015). REPTree uses the
regression tree logic and creates multiple trees in
different iterations. The best one is then selected to
be considered representative. The tree is pruned
using mean square error. It is a fast algorithm which
uses information gain as the splitting criterion.
Missing attributes are dealt with using C4.5’s
fractional instances (Kalmegh, 2015).
On MATLAB®, however, the fit tree function
uses the Classification And Regression Tree
(CART) algorithm, which creates a large tree and
then prunes it to a certain size based on cross-
validation estimate of error (Loh, 2014).
2.3.2 Artificial Neural Networks
The brain is basically constituted of neurons
interconnected with axons and dendrites. It learns by
adjusting these connections (Aguiar-Pulido et al.,
2013). Since it is a complex, nonlinear and parallel
processing system, it is interesting to simulate this
capacity (Greer and Khan, 2004).
ANNs resemble the brain in two aspects. First,
knowledge is acquired through learning. Second,
strong interneural connections (synaptic weights) are
used to store knowledge (Greer and Khan, 2004).
These ANNs are made of nodes called neurons,
and directed links which measure signal importance
using a weight factor. These values adapt based on
information processed during learning (Aguiar-
Pulido et al., 2013). A ANN is developed in three
stages, design, training and validation. Designing
involves network architecture selection (number of
neurons and layers). The optimal selection is usually
subjective and requires trial and error (Gamito and
Crawford, 2004).
Input data are analogous to independent variable
(x) and output data to dependent variable (y). When
training starts, connection weights are small. Hence,
the output values are arbitrary and errors are high. In
supervised learning, the output given by the network
is compared to the expected output. A training
algorithm adjusts the weights based on the
calculated error from the two outputs (Gamito and
Crawford, 2004).
During training, instances with known output are
presented to the ANN sequential and repeatedly,
constituting epochs or iterations. The algorithm
adjusts the weights and, with time, a matrix is
generated, which presents the training values that
minimize error (Gamito and Crawford, 2004).
In the present study, the ANNs were
programmed on MATLAB® using the pattern
recognition function. The data were divided for
training (70%) and for testing (30%). Then, ANNs
with different architectures were trained, analyzing
the number of layers (one or two) and the number of
neurons in each layer (from one to 100 for the first
layer and one to 15 in the second one), as well as the
number of trainings an ANN should perform. From
all the generated ANNs, the one which presented the
lowest error in classification was saved.
2.4 Data Analysis
In order to analyze the results, 10 fold cross
validation, ROC curve and confusion matrices were
used in the present study.
3 RESULTS AND DISCUSSION
3.1 Decision Trees
On Weka®, the J48 algorithm was used with all
native configurations. Figure 1 shows the tree
generated by J48, it’s worth noting that ’t’ and ’n’
stand for tumoral and normal, respectively. It used
four different genes to classify the instances (TSLP,
FYN, PSENEN and RABIF). Observing the
numbers at the end of each node, it is possible to
notice that TSLP and FYN together were able to
classify most part of the instances.
TSLP (Thymic Stromal Lymphopoietin) is a
member of the cytokine IL-2 family and a distant
paralogue of IL-7. The murine gene was discovered
in thymic stromal cell lineages that supported B cell
development. Just like IL-7, this gene can stimulate
thymocytes to promote B cell lymphopoiesis (Roan
et al., 2012). A homologue of this protein was
identified in humans through computational
methods. In a similar way, it was possible to isolate
Comparative Study on Data Mining Techniques Applied to Breast Cancer Gene Expression Profiles
171

Figure 1: Decision tree created on Weka through J48
algorithm.
a receptor for the TSLP. Besides, several cell types
respond to this protein, including innate immune
system cells, which is evidenced by the possibility
of cytokine production in mast cells, NK and
eosinophils. Interestingly, the role of TSLP is quite
complicated. Some studies found a promoting role in
tumor development. Others, on the other hand, a
suppressing role. Therefore, the importance of this
gene as a possible target for cancer therapy is
evidenced (LoKuan and Ziegler, 2014).
The FYN gene, on the other hand, is located in
chromosome 6q21. It is a member of the SRC
family, originally identified in 1986 as SYN or SLK.
It is located primarily in the plasma membrane,
where if phosphorylates tyrosine residues in targets
involved in different signaling paths. Its biological
roles are quite diverse. In the past, a lot was focused
on its immunologic and neurologic functions.
However, this gene is also mitogenic signaling
mediator and regulator of the cell cycle, growing and
proliferation, integrin mediated interactions and cell
adhesion. High expression levels have been linked to
morphological transformations in normal cells. For
example, high expression of this gene in NIH 3T3
fibroblasts showed a phenotype similar to that of
cancer, with increased anchorage independent
growth and prominent morphological alterations.
This gene is over expressed in several cancer types,
including glioblastomas, neck and head squamous
cell carcinoma, and melanoma. However, the role
behind this increased expression has not been well
defined yet (Saito et al., 2010).
Kinases of the SRC family (SFKs) were among
the first kinases to be discovered. The family is
constituted of 11 members, 8 of which have already
been studied. Some, like c-Src has been intensely
studied when it comes to it relationship with cancer
biology, particularly as a molecule of vital
importance in tumor development, progression and
resistance to therapeutic agents. For the last decade,
the involvement of other members of the family, like
Fyn, in several aspects of cancer biology has become
more apparent (Elias and Ditzel, 2015).
Fyn is located in the most internal layer of the
plasma membrane, attached to myristic and palmitic
acids. The activity of this protein is regulated by
intermolecular interactions influenced by tyrosine
phosphorylation and dephosphorylation. Fyn
activation leads to tyrosine phosphorylation in
different targets, like focal activation kinase (FAK)
and anti estrogen resistance protein 1 (BCAR1)
(Elias and Ditzel, 2015).
This gene has several molecular functions,
including cell growth regulation, survival, adhesion,
cytoskeleton remodeling, motility, axon direction,
synaptic function, myelination in the central nervous
system, placket activation and T cell receptor
signaling (Elias and Ditzel, 2015).
Fyn is also involved in several pathogenic
aspects of different types of cancers, with tumor
promoting effects, proliferation promotion,
migration and prostate tumor cell invasion, cell
death inhibition and mesenchymal epithelial
transition. Again, the importance of studying this
gene as a possible target for cancer therapy get
highlighted (Elias and Ditzel, 2015).
However, neither PSENEN or RABIF could be
specifically found linked to cancer development.
Figure 2, on the other hand, shows the decision
tree built by REPTree. On this tree, only one gene
was used to classify the instances, MMP11.
The matrix metalloproteinases (MMP) are
important components of the tumor stroma. They
regulate and shape the tumor microenvironment, its
expression, and activation, all of which are increased
in most human cancers when compared to levels
found in normal tissues. The MMP11 was isolated
as a gene associated to breast cancer, and it is
expressed in most primary invasive carcinomas, in
several of its metastasis and, although more rarely,
in sarcomas and other epithelial malignancies. It is
worth noting that it is a protein that is barely even
expressed in normal adult tissues (Peruzzi et al.,
2009).
Figure 2: Decision tree created on Weka through the
REPTree algorithm.
Peruzzi and coworkers (2009) analyzed if
MMP11 was a relevant target for human cancer
BIOINFORMATICS 2017 - 8th International Conference on Bioinformatics Models, Methods and Algorithms
172
through analysis of microarray data of stomach,
kidney, colon, lung and breast cancers. The
abundance of this protein in these tissues was
compared to normal tissue values from the same
type, which aimed at analyzing the differential
expression of this particular protein. The authors
found out that MMP11 is more expressed in all
types of tumor when compared to normal tissue,
which highlights the role of this particular gene as a
target for cancer therapy.
Although on MATLAB® DT of different
complexities were testes, the simplest one was
already able to classify the instances with a good
percentage (98%) of correct classifications.
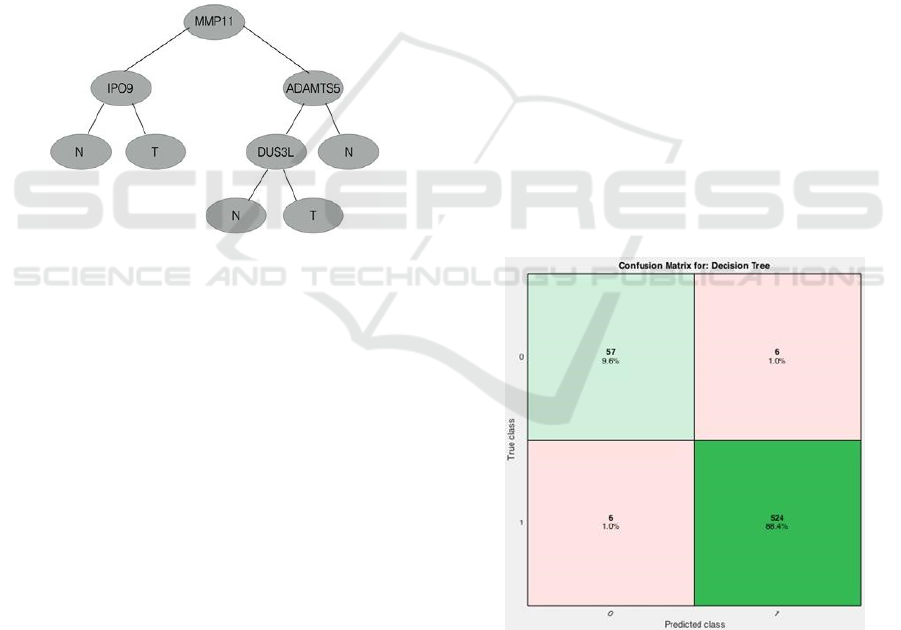
The genes identified on MATLAB® are different
from those found on Weka® (MMP11, IPO9,
ADAMTS5, DUS3L) except for MMP11. This tree
can be observed in Figure 3.
Figure 3: Decision tree created on MATLAB®.
Members of the ADAMTS family can be
classified in different groups, which are based on its
functions. Interestingly, it is worth mentioning that
on recent years several studies have arisen about the
role of this family in angiogenesis and cancer.
Several members were linked to angiogenic
regulation, which were placed on the list of
angiogenesis inhibitors. However, this mechanism is
not shared by all members of this family (Kumar et
al., 2012).
ADAMTS5, is one of the most studied
metalloproeinases from this family due to its role in
cartilage degradation in arthritis. The human gene is
located in chromosome 21q21.3. Small chemical
inhibitors of this metalloproteinase were identified
based on its catalytic domain structure. Recent
studies evidenced another role of this protein,
involved in the development of other conditions
such as cancer. There is a crescent number of
publications about the regulation of this gene
expression levels in the progression of malignant
tumors, suggesting that it may act as a suppressor in
some types of cancer. A decrease in the mRNA
levels for this gene was identified in prostate cancer
besides head and neck squamous cell carcinoma
when compared to normal levels. Moreover, in
breast cancer tissue, this protein levels are low when
compared to non-neoplastic tissue (Kumar et al.,
2012).
In a study conducted by Porter and coworkers
(2004), this gene was shown as being repressed in
breast cancer. The authors compared the mRNA
level of all ADAMTS genes in malignant breast
tumors and non neoplastic breast tissue. They were
able to show that ADAMTS5 was repressed in this
type of cancer. Furthermore, although there is a low
ADAMTS5 expression in prostate cancer cell
lineages, normal cells from the prostate estroma
present high levels of ADMATS5 expression. It was
also documented that this gene would be
hypermethylated in colorectal cancer. Therefore, a
pattern arises which suggest a supressor role for this
gene (Kumar et al., 2012).
Nevertheless, other publications suggest that
ADAMTS5 would be over expressed in
glioblastoma, and that it could contribute to glial cell
invasion. Hence, ADAMTS5 has different roles in
different types of cancer which depend on substrate
availability and its antiangiogenic activity (Kumar et
al., 2012).
The gene IPO9 could not be directly associated
with the development of breast cancer.
Figure 4: Confusion matrix generated on MATLAB® for
the corresponding decision tree. 98% of the samples were
correctly classified by the tree (sum of the diagonals). 12
out of 593 samples were misclassified.
The confusion matrix shows the predicted class
versus the real class to which a certain patient
belongs. In this matrix, it is possible to observe the
Comparative Study on Data Mining Techniques Applied to Breast Cancer Gene Expression Profiles
173

percentage of correctly classified instances. On the
matrix obtained on MATLAB®, the classifier was
able to correctly classify 98% of the instances
(Figure 4).
As it was mentioned previously, the quality of a
certain classifier can be expressed in terms of a ROC
curve, as can be seen in Figure 5. Two aspects can
be seen in this curve, the position of the current
classifier and the area under the curve. These values
are better the closer they get to 1. In this case, the
area under the curve was 0.947574 and the classifier
can be found in a point of coordinates (0.0113208
0.904762).
Figure 5: ROC curve for the decision tree on MATLAB®.
The point represents the efficiency of the classifier, which
is calculated from the division of the false positive rate
and the true positive rate.
3.2 Artificial Neural Networks
Among the several ANNs generated on MATLAB®,
the one selected as the best was the one which
presented the lowest global error in the classification
process. This structure of this particular ANN can be
seen in Figure 6.
An error histogram plot was created for this
particular ANN (Figure 7) on which it can be seen
that from all the patients used for testing, only 2 had
an error different from -0.00525.
Figure 7: Error histogram with the error values for the
best.
Moreover, the classification made by the ANN
was also evaluated by means of a confusion matrix
(Figure 8). In the picture, it can be seen that 98,9%
of the samples were correctly classified. Only one
patient from each group was misclassify.
Figure 8: Confusion matrix calculated for the ANN which
presented the lowest global error.
Figure 6: Structure of the ANN which presented the lowest global error among all the different networks created on
MATLAB®.
BIOINFORMATICS 2017 - 8th International Conference on Bioinformatics Models, Methods and Algorithms
174

4 CONCLUSIONS
The use of both programs for implementation of
DTs showed diversity in the genes selected by the
same method, with at least TSLP, FYN and MMP11
being directly linked to cancer. Moreover, the ANNs
presented good classification capabilities, with the
one selected presenting very low errors (-0.00525).
For both methods, confusion matrices showed
correct classification of at least 98% of instances.
Although there are some divergent results especially
when it comes to the DT created in the two different
pieces of software, the results are coherent to what
was set as objective on the present study (i.e., using
of data mining techniques for discovery of genes
associated with breast cancer development).
REFERENCES
Aguiar-Pulido, V., et al. 2013. Exploring patterns of
epigenetic information with data mining techniques.
Curr Pharm Des, 19, 779-89.
Ahmad, P., et al. 2015. Techniques of Data Mining In
Healthcare: A Review. International Journal of
Computer Applications, 120, 38-50.
BRASIL. Instituto Nacional do Cancer (INCA). 2016. O
que é o cancer? Available at
http://www1.inca.gov.br/conteudo_view.asp?id=322.
Accessed: 27 March 2016.
Desantis, C.E., et al. 2015. International Variation in
Female Breast Cancer Incidence and Mortality Rates.
Cancer Epidemiol Biomarkers Prev, 24, 1495-506.
Elias, D., Ditzel, H.J. 2015. Fyn is an important molecule
in cancer pathogenesis and drug resistance.
Pharmacological Research, 100, 250-254.
EUA. The Cancer Genome Atlas. National Institute of
Health. 2016. About TCGA. Available at
<http://cancergenome.nih.gov/abouttcga>. Accessed
27 March 2016.
Ferlay, J., et al. 2015. Cancer incidence and mortality
worldwide: sources, methods and major patterns in
GLOBOCAN 2012. Int J Cancer, 136, E359-86.
Gamito, E.J., Crawford, E.D. 2004. Artificial neural
networks for predictive modeling in prostate cancer.
Curr Oncol Rep, 6, 216-21.
Greer, B.T., Khan, J. 2004. Diagnostic classification of
cancer using DNA microarrays and artificial
intelligence. Ann N Y Acad Sci, 1020, 49-66.
Hall, M.A., et al. 2009. The WEKA Data Mining
Software: An Update. SIGKDD Explorations 11, 1.
Kalmegh, S. 2015. Analysis of WEKA Data Mining
Algorithm REPTree, Simple Cart and RandomTree for
Classification of Indian News. IJISET - International
Journal of Innovative Science, Engineering &
Technology, 2.
Kingsford, C., Salzberg, S. L. 2008. What are decision
trees? Nat Biotechnol, 26, 1011-3.
Kumar, S., et al. 2012. Emerging Roles of ADAMTSs in
Angiogenesis and Cancer. Cancers, 4, 1252-1299.
Liu, B., et al. 2004. A combinational feature selection and
ensemble neural network method for classification of
gene expression data. BMC Bioinformatics, 5, 136.
Loh, W.-Y. 2014. Fifty Years of Classification and
Regression Trees. International Statistical Review, 82,
329-348.
Lokuan, E., Ziegler, S.F. 2014. Thymic Stromal
Lymphopoietin (TSLP) and Cancer. Journal of
immunology (Baltimore, Md. : 1950), 193, 4283-4288.
Peruzzi, D., et al. 2009. MMP11: A Novel Target Antigen
for Cancer Immunotherapy. Clinical Cancer Research,
15, 4104-4113.
Podgorelec, V., et al. 2002. Decision trees: an overview
and their use in medicine. J Med Syst, 26, 445-63.
Porter, S., et al. 2004. Dysregulated Expression of
Adamalysin-Thrombospondin Genes in Human Breast
Carcinoma. Clinical Cancer Research, 10, 2429-2440.
Roan, F., et al. 2012. The multiple facets of thymic
stromal lymphopoietin (TSLP) during allergic
inflammation and beyond. Journal of Leukocyte
Biology, 91, 877-886.
Sa'di, S., et al. 2015. Comparison of Data Mining
Algorithms in the Diagnosis of Type Ii Diabetes.
International Journal on Computational Science &
Applications, 5, 1-12.
Saito, Y.D., et al. 2010. Fyn: a novel molecular target in
prostate cancer. Cancer, 116, 1629-1637.
Shah, S., Kusiak, A. 2007. Cancer gene search with data-
mining and genetic algorithms. Comput Biol Med, 37,
251-61.
Tseng, W.T., et al. 2015. The application of data mining
techniques to oral cancer prognosis. J Med Syst, 39,
59.
Witten, I.H., et al., 2011. Data Mining Practical Machine
Learning Tools and Techniques, 3rd ed. Morgan
Kaufmann Publishers, USA.
Comparative Study on Data Mining Techniques Applied to Breast Cancer Gene Expression Profiles
175
