
Importance of Sequence Design Methods Considering Hybridization
Kinetics for in vivo DNA Computers
Toshihiro Kojima and Akira Suyama
Life Sciences and Institute of Physics, University of Tokyo, 3-8-1, 153-8902, Komaba, Meguro-Ku, Tokyo, Japan
Keywords: Sequence Design, DNA Computer, Hybridization Kinetics.
Abstract: A DNA computer is a DNA-based synthetic system inspired by biology. One of the goals of DNA computer
research is to develop an in vivo DNA computer, which can function within living cells through non-
destructively processing intracellular signals under isothermal conditions. DNA computers working in
isothermal conditions need a set of nucleotide sequences satisfying a kinetic condition in addition to the
thermodynamic conditions considered previously, because the progress of computation under isothermal
conditions is often dominated by the rate of nucleic acid hybridization reactions. We thus developed a
method to predict the hybridization reaction rate from nucleotide sequences and have demonstrated
experimentally the importance of hybridization reaction rates and the usefulness of our method. The present
method is general and can be used to develop any hybridization-based DNA/RNA system such as DNA
computers, DNA sensors, DNA nanostructures, and nucleic acid drugs, working in isothermal conditions.
1 DEOXYRIBONUCLEIC ACID
(DNA) BASED SYNTHETIC
SYSTEMS
Recent advances in DNA nanotechnology have
allowed the development of several types of DNA-
based synthetic systems (Stulz et al., 2011). A DNA
computer (Adleman, 1994), for instance, is one such
DNA-based synthetic system that can perform
computations. DNA computers employ nucleic acid
reactions, such as hybridization, strand exchange,
and enzymatic strand synthesis and cleavage
reactions to process input information and produces
output results. Hybridization reactions play an
important role in DNA computers and DNA-based
synthetic systems as they enable the systems to
possess highly programmable attributes.
2 in vivo DNA COMPUTERS
One of the goals of DNA computer research is to
develop an in vivo DNA computer, which can
function within living cells (Benenson, 2012;
Hemphill and Deiters, 2013). In vivo DNA
computers are expected to be widely used in fields
ranging from basic to applied biology and medicine.
For instance, an in vivo DNA computer can be used
as a non-destructive measuring instrument. It can
collect information on cell conditions from mRNAs
transcribed in a cell, and can report the information
outside the cell by enclosing it in vesicles such as
exosomes. An in vivo DNA computer can also be
used to develop intelligent drugs. This computer can
use the information it has collected to produce RNA
and protein molecules as drugs to treat unfavorable
cell conditions or induce differentiation of cells to
form organs.
Fuzzy logic (Kosko and Isaka, 1993) may
provide a theoretical basis for the operation of in
vivo DNA computers. Values of data in vivo DNA
computers can collect in living cells are in fact
fuzzy. In addition, molecular reaction circuits of in
vivo DNA computers cannot perform sufficiently
precise operations, unlike electronic circuits. Thus,
implementation of fuzzy logic may be important for
the operation of in vivo DNA computers.
Certain evolutionary systems may be
successfully combined with in vivo DNA computers
to more closely mimic the process of natural
selection. For instance, an in vivo DNA computer
could automate iterative cycles of mutagenesis,
selection, and amplification processes employed in
the directed evolution (or evolutionary molecular
248
Kojima T. and Suyama A.
Importance of Sequence Design Methods Considering Hybridization Kinetics for in vivo DNA Computers.
DOI: 10.5220/0006246902480252
In Proceedings of the 10th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2017), pages 248-252
ISBN: 978-989-758-212-7
Copyright
c
2017 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
engineering) method (Packer and Liu, 2015) to
simulate the evolution of proteins or nucleic acids
toward a user-defined goal. Automation allows the
user to optimize the method.
Figure 1: Central dogma of molecular biology.
3 REVERSE-TRANSCRIPTION-
AND-TRANSCRIPTION-BASED
AUTONOMOUS COMPUTING
SYSTEM (RTRACS)
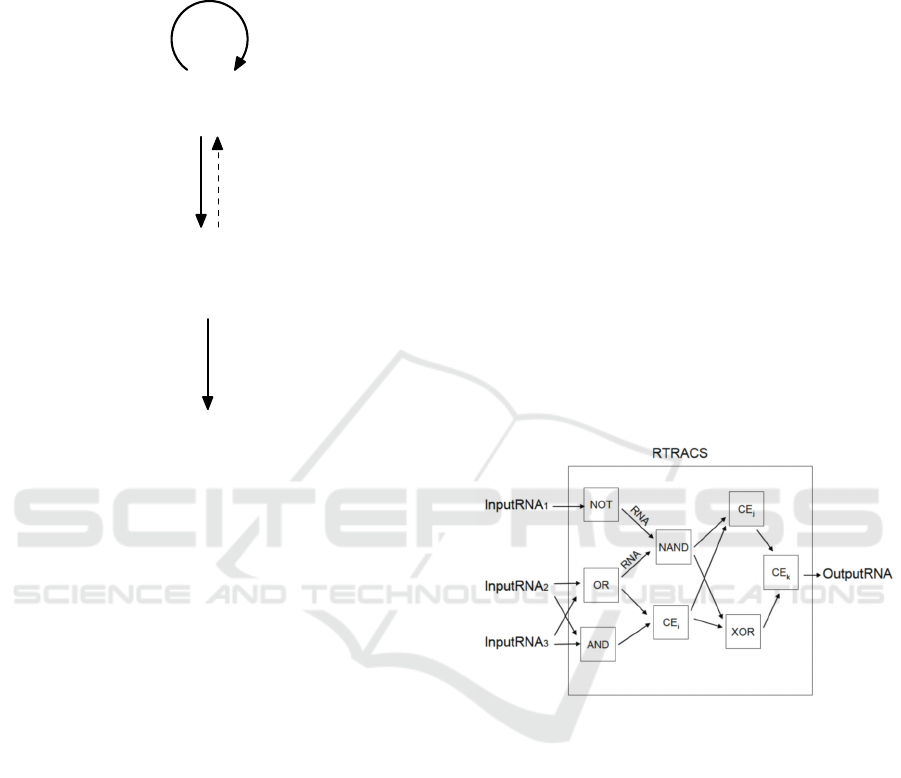
RTRACS is a modular DNA computer that works
under isothermal conditions using DNA as a
program and RNA as a variable (Nitta and Suyama,
2004; Takinoue et al., 2008). RTRACS provides a
promising framework for in vivo DNA computers,
because the idea of RTRACS was inspired by the
central dogma of molecular biology. The dogma
defines the flow of genetic information within a
biological system (Fig. 1). Genomic DNA storing
genetic information in the form of a DNA sequence
is transcribed onto a messenger RNA (mRNA),
which is then translated into a protein to assemble
replicated systems. Considering information stored
in genomic DNA as a source program and small
pieces of the information transferred into mRNA as
variables, the information flow within a biological
system and that within a computer appear to be
homologous. This implies that a biological system
is itself a DNA computer that uses DNA as a
program and RNA as a variable. RTRACS uses
DNA and RNA in a manner similar to a biological
system. Therefore, RTRACS is highly compatible
with a biological system, and can provide a suitable
framework for in vivo DNA computers.
RTRACS performs computations using the
network or circuit of modularized computational
elements called function modules. In the network,
function modules are connected together by RNA
molecules (Fig. 2). An RTRACS program is written
using the network structure of function modules. A
function module accepts input RNA molecules and
returns output RNA molecules produced through
molecular reactions that include DNA-DNA
hybridizations, DNA-RNA hybridizations, and
enzymatic reactions with reverse transcriptase,
RNase H, and RNA polymerase. The input and
output RNA sequences contain not only the values
for input and output data but also information
regarding the connection of function modules. The
RNA sequences consist of code word sequences
(orthonormal sequences) with high sequence
specificity.
Figure 2: Network of function modules used in RTRACS
to perform computation.
4 RTRACS-BASED in vivo DNA
COMPUTER
We describe here, an example of a synthetic genetic
circuit constructed using RTRACS function
modules. The synthetic circuit is compatible with
endogenous genetic circuits, namely, genetic circuits
within a cell. Thus, the synthetic gene circuit can
work as an in vivo DNA computer, which measures
cellular conditions and applies appropriate action to
the cell in response to these conditions.
The synthetic genetic circuit has two input
mRNAs and one output mRNA translated into a
target protein (Fig. 3a) and works as follows. First,
DNA
RNA
protein
replicati on
transcription
translation
reverse
transcription
Importance of Sequence Design Methods Considering Hybridization Kinetics for in vivo DNA Computers
249
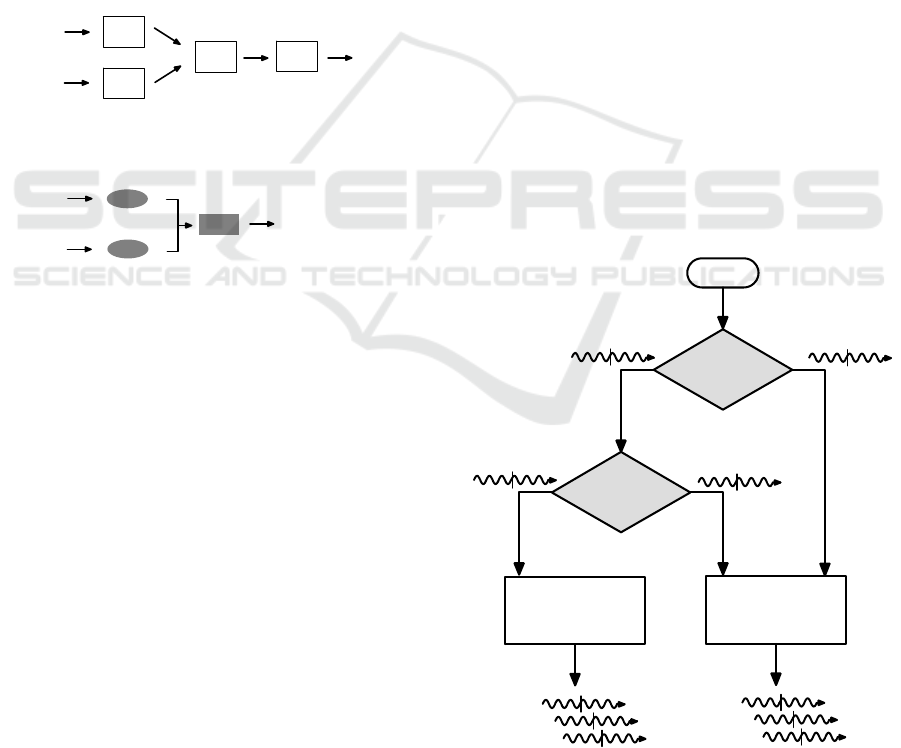
two input mRNAs, X and Y, are encoded as RNA
sequences used in RTRACS. Second, the coded
RNAs are processed by the function module that
performs the AND operation, and an output RNA is
produced according to the value of the operation
result. Third, the produced RNA is decoded into
mRNA of protein Z. Finally, the mRNA is translated
into protein Z. Unlike the natural genetic circuit
inherently present in a cell (Fig. 3b), the synthetic
genetic circuit (Fig. 3a) has an advantage that the
input mRNA molecules are not necessarily the
sequences of transcription factors. In addition, any
protein can be produced as the output. Preliminary
experiments using green fluorescence protein as the
output protein demonstrated that the synthetic
genetic circuit functions within a cell-free protein
synthesis environment.
Figure 3: Regulation of gene expression with transcription
factors. (a) A synthetic mechanism constructed using
RTRACS. (b) A natural mechanism widely employed for
genetic circuits in living cells.
5 SEQUENCE DESIGN FOR in
vivo DNA COMPUTERS
DNA computers need a set of nucleotide sequences
of high hybridization specificity. A reliable design
method for such a sequence set usually adopts the
following three thermodynamic conditions: presence
of normality, orthogonality, and prevention of stable
secondary self-folding. Normality implies that all
sequences within the set have similar melting
temperatures, while orthogonality indicates that any
two sequences in the set, except sequences
complementary to each other, do not hybridize into a
stable double strand with comparable melting
temperature to a double strand formed between
complementary strands. The prevention of stable
secondary self-folding helps sequences take double-
stranded forms with complementary strands rather
than maintain their single-stranded form.
DNA computers working in isothermal
conditions, such as RTRACS and in vivo DNA
computers, need a set of nucleotide sequences
satisfying a kinetic condition in addition to the three
thermodynamic conditions. Namely, all
hybridization reaction rates are required to be
adequate for reliable computation. The progress of
computation under isothermal conditions is often
dominated by the rate of nucleic acid hybridization
reactions. Therefore, various undesirable problems
would occur in computation if the hybridization
reaction rates are not appropriate. For example, a
computation is not completed within the expected
timeline if the computation includes a very slow
hybridization step. Timing problems tend to occur in
parallel computing if the speed of computation
differs largely between processes.
To ensure that the design of nucleotide sequence
sets satisfies the kinetic condition, we developed a
method to predict the rate of hybridization reactions
involved in DNA computation from nucleotide
sequences. The method predicts the rate based on the
nucleation-zippering model of complementary
strand hybridization and can be applied not only to
DNA-DNA hybridization reactions but also to
DNA-RNA and RNA-RNA hybridization reactions.
Figure 4: Schematic diagram of computational processes
in logic gate modules.
mRNA X
mRNA Y
protein Z
ENC
ENC
AND
DEC
mRNA X
mRNA Y
transcription
factor X
transcription
factor Y
gene
protein Z
(b)
(a)
Start
X'
Z
XorX'?
YorY'?
output Z output Z'
Z'
X
Y'
Y
BIOSIGNALS 2017 - 10th International Conference on Bio-inspired Systems and Signal Processing
250

Figure 5: Output production rates of LGM1 and LGM2. (a) Z and (b) Z´ output of LGM1. (c) Z and (d) Z´ output of LGM2.
The rate is defined as the speed at which a change in the intensity of fluorescence emitted from output RNA occurs. (e) The
input-output relation (truth table) for LGM and line types of the input combinations. The results were obtained as follows.
The reaction mixtures contained 100 mM primer DNA cx that specifically binds to input X, 10 mM primer DNA cx´ that
specifically binds to input X´, 2 mM each of converter DNAs transcribing output Z and Z´, 100 mM each of input RNAs
corresponding to the specified input combination, 250 mM each of 2´-O-methyl molecular beacons with FAM and Cy5
dyes specifically binding to output Z and Z´, respectively, 0.3 unit/μL AMV Reverse Transcriptase (Promega), 1 unit/μL
Hybridase Thermostable RNase H (Epicentre), Thermo T7 RNA polymerase (Toyobo), 5 mM DTT, 0.15 mM of each
dNTP, and 1 mM of each NTP in a 20 μL reaction buffer (40 mM Tris-HCl [pH 8.0], 50 mM NaCl, 9.6 mM MgCl
2
). The
mixtures were prepared on ice and immediately incubated at 50°C to measure the fluorescence intensity of FAM (Z output)
and that of Cy5 (Z´ output) once every 2 minutes using a real time polymerase chain reaction detection system, CFX96
(Bio-Rad).
The method was applied to design nucleotide
sequence sets for logic gate modules (LGMs) of
RTRACS. LGM can perform various logical
operations including AND, OR, NAND, NOR, INH,
and NINH (Kan et al., 2014). A schematic diagram
of the computational processes forming LGM is
shown in Fig. 5. LGM accepts four combinations of
input RNA molecules: (X, Y), (X, Y´), (X´, Y), and
(X´, Y´). The output RNA molecule Z is produced
only if the input combination is (X, Y). The output
RNA molecule Z´ is produced if the combination is
one of the other three. There are DNA-RNA
hybridization reactions in the decision process of X
or X´, and Y or Y´. We designed two sets of
nucleotide sequences, one for LGM1 and the other
for LGM2. The sequence set for LGM1 was
designed using only the thermodynamic conditions.
In contrast, the set for LGM2 was designed using
both, kinetic and thermodynamic conditions. After
applying the kinetic condition to nucleotide
sequences in the set for LGM1, those sequences that
did not satisfy the kinetic condition were replaced
with those satisfying both, kinetic and
thermodynamic conditions to generate the sequence
set for LGM2.
Experimental results of the operation of LGM1
and LGM2 agreed with the predicted kinetic
properties of their sequences. LGM1 did not work
correctly and produced no output RNA Z for the
input combination (X, Y), while LGM2 correctly
(a) (b)
(c) (d)
(e)
0
50
100
150
0204060
ProductionRate(a.u./min)
Time(min)
0
50
100
150
0204060
ProductionRate(a.u./min)
Time(min)
0
50
100
150
0204060
ProductionRate(a.u./min)
Time(min)
0
50
100
150
0204060
ProductionRate(a.u./min)
Time(min)
In Out
XY Z
XY′ Z′
X′ YZ′
X′ Y′ Z′
‐‐ ‐
Importance of Sequence Design Methods Considering Hybridization Kinetics for in vivo DNA Computers
251

produced output RNA Z according to the truth table
of LGM (Fig. 6a, c). As for output RNA Z´, LGM1
and LGM2 both, correctly produced the output (Fig.
6b, d). These results were consistent with the
hybridization rate prediction, which stated that the
hybridization reaction rate related to the decision
process of Y in LGM1 only was too slow to produce
output RNA Z promptly. These results demonstrate
the importance of predicting hybridization reaction
rates for the development of LGMs and usefulness
of the present kinetic method.
Our kinetic method to predict the hybridization
reaction rate from nucleotide sequences is general
and its application is not limited to the development
of LGMs of RTRACS. The method can be used to
develop any hybridization-based DNA/RNA system
such as DNA computers, DNA sensors, DNA
nanostructures, and nucleic acid drugs, especially
working in isothermal conditions. The method is
also useful in other areas of biological research, such
as identification of non-coding RNA functions and
understanding their mechanism.
ACKNOWLEDGEMENTS
This work was supported by Grant-in-Aid for
Scientific Research on Innovative Areas [23119007]
to A.S. from the Ministry of Education, Culture,
Sports, Science, and Technology, Japan. H.K.
acknowledges support from the Japan Society for the
Promotion of Science through Program for Leading
Graduate Schools (ALPS).
REFERENCES
Adleman, L. M. 1994. Molecular computation of solutions
to combinatorial problems. Nature, 369(40), 1021-
1024.
Benenson, Y. 2012. Biomolecular computing systems:
principles, progress and potential. Nature Reviews
Genetics, 13(7), 455-468.
Hemphill, J., Deiters, A. 2013. DNA computation in
mammalian cells: microRNA logic operations.
Journal of the American Chemical Society, 135(28),
10512-10518.
Kan, A., Sakai, Y., Shohda, K. I., & Suyama, A. 2014. A
DNA based molecular logic gate capable of a variety of
logical operations. Natural Computing, 13(4), 573-581.
Kosko, B., & Isaka, S. 1993. Fuzzy logic. Scientific
American, 269(1), 76-81.
Nitta, N., & Suyama, A. 2004. Autonomous biomolecular
computer modeled after retroviral replication. Lecture
Notes in Computer Science, 2943, 203-212.
Packer, M. S., & Liu, D. R. 2015. Methods for the directed
evolution of proteins. Nature Reviews Genetics, 16(7),
379-394.
Stulz, E., Clever, G., Shionoya, M., & Mao, C. 2011.
DNA in a modern world. Chemical Society Reviews,
40(12), 5633-5635.
Takinoue, M., Kiga, D., Shohda, K. I., & Suyama, A.
2008. Experiments and simulation models of a basic
computation element of an autonomous molecular
computing system. Physical Review E, 78(4), 041921.
BIOSIGNALS 2017 - 10th International Conference on Bio-inspired Systems and Signal Processing
252
