
Multi-scale Convolutional Neural Networks for the Prediction of
Human-virus Protein Interactions
Xiaodi Yang
1a
, Ziding Zhang
1,* b
and Stefan Wuchty
2,3* c
1
State Key Laboratory of Agrobiotechnology, College of Biological Sciences, China Agricultural University, Beijing
100193, China
2
Dept. of Computer Science, University of Miami, Miami FL, 33146, U.S.A.
3
Dept. of Biology, University of Miami, Miami FL, 33146, U.S.A.
Keywords: Human-virus PPI, Prediction, Deep Learning, PSSM, CNN, Transfer Learning.
Abstract: Allowing the prediction of human-virus protein-protein interactions (PPI), our algorithm is based on a
Siamese Convolutional Neural Network architecture (CNN), accounting for pre-acquired protein evolutionary
profiles (i.e. PSSM) as input. In combinations with a multilayer perceptron, we evaluate our model on a variety
of human-virus PPI datasets and compare its results with traditional machine learning frameworks, a deep
learning architecture and several other human-virus PPI prediction methods, showing superior performance.
Furthermore, we propose two transfer learning methods, allowing the reliable prediction of interactions in
cross-viral settings, where we train our system with PPIs in a source human-virus domain and predict
interactions in a target human-virus domain. Notable, we observed that our transfer learning approaches
allowed the reliable prediction of PPIs in relatively less investigated human-virus domains, such as Dengue,
Zika and SARS-CoV-2.
1 INTRODUCTION
Deep learning as a branch of machine learning
represents information through artificial neural
network modules, which share similar properties with
neural modules in the brain (Kriegeskorte and
Douglas, 2018; Yamins and DiCarlo, 2016). In the
past decade, applications of deep learning approaches
demonstrated improved performance in many fields
(e.g. biomedicine, image, speech recognition, etc)
(Karimi et al., 2019; Pospisil et al., 2018; Sainath et
al., 2015). In particular, convolutional neural
networks (CNN) (Hashemifar et al., 2018) and
recurrent neural networks (RNN) (Zhang et al., 2016)
automatically capture local features in images as well
as preserve contextualized/long-term ordering
information in sequence data. In addition, many
recent studies adopt a Siamese network architecture
based on CNN or RNN to capture mutual influence
between two individual inputs (Chen et al., 2019;
Hashemifar et al., 2018).
a
https://orcid.org/0000-0002-3229-5865
b
https://orcid.org/0000-0002-9296-571X
c
https://orcid.org/0000-0001-8916-6522
In general, traditional machine learning/deep
learning can only perform well, if training and test
sets are cut from the same feature space, ensuring
similar statistical distributions of feature values.
(Shao et al., 2015). While the rigid application of a
trained model on data sets with different distributions
usually perform poorly, transfer learning methods
utilize prior knowledge from a ‘source’ to train in a
‘target’ task domain (Chang et al., 2018; Shao et al.,
2015). In particular, transfer learning approaches
have been successfully applied to tackle problems in
many fields, such as medical imaging (Cheplygina et
al., 2019), biomedicine (Taroni et al., 2019), and
visual categorization (Shao et al., 2015). A regular
phenomenon appears in various training objectives
(Le et al., 2011; Lee et al., 2009) in that the first layers
of deep neutral networks (DNN) usually capture
standard features of the training data, providing a
foundation for transfer learning. Specifically, a deep
neural network can be trained on a source task,
establishing the parameters of the first layers.
Yang, X., Zhang, Z. and Wuchty, S.
Multi-scale Convolutional Neural Networks for the Prediction of Human-virus Protein Interactions.
DOI: 10.5220/0010185300410048
In Proceedings of the 13th International Conference on Agents and Artificial Intelligence (ICAART 2021) - Volume 2, pages 41-48
ISBN: 978-989-758-484-8
Copyright
c
2021 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
41
Subsequently, parameters of late layers are trained on
the target task. Depending on the size of the target
dataset and number of parameters of the DNN, first
layers of the target DNN can either remain unchanged
during training on the new dataset (i.e. frozen), or
fine-tuned towards the new task, indicating a balance
between specificity and generality of derived prior
knowledge.
Here, we propose a framework to predict
interactions between virus and human proteins that is
based on a Siamese Convolutional Neural Network
architecture (CNN), accounting for pre-acquired
protein evolutionary profiles (i.e. PSSM) as protein
sequence input. In combination with a multilayer
perceptron (MLP), we assess the prediction
performance of our model on different human-virus
PPI datasets, outperforming other prediction
frameworks. Allowing to predict interactions in a
target domain of human-virus interactions, we
propose two types of transfer learning methods where
we freeze/fine-tune weights learned in the Siamese
CNN. Notably, the transfer of prior knowledge
learned from a large-scale human-virus PPI dataset
allowed the reliable prediction of PPIs between
human and proteins of less well investigated viruses
such as Dengue, Zika and SARS-CoV-2.
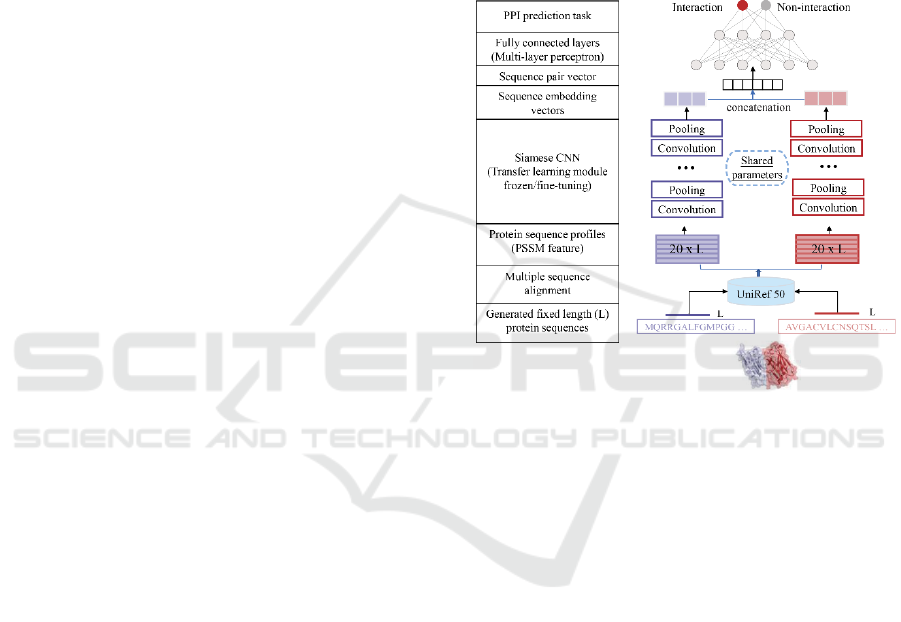
2 MATERIALS AND METHODS
2.1 Deep Neural Networks Framework
Representing interactions between human and viral
proteins through their amino-acid sequences, we
introduce an end-to-end deep neural network
framework, called a Siamese-based CNN that
consists of a pre-acquired protein sequence profile
module, a Siamese CNN module and a prediction
module (Fig. 1). In particular, the Siamese
architecture of the CNN module allows us to account
for residual relationships between interacting viral
and human protein sequences through protein
sequence profiles (i.e. PSSM) that capture
evolutionary relationships between proteins. Such
latent protein profile representations of interacting
protein pairs are fed to the Siamese CNN module to
generate respective high-dimensional sequence
embeddings. Finally, output embeddings of two
proteins are combined to form a sequence pair vector
as the input of a multilayer perceptron (MLP) with an
appropriate loss function to predict the
presence/absence of an interaction between a viral
and a human protein.
2.1.1 Pre-acquired Protein Sequence Profile
Module
For each protein sequence with variable lengths, we
generate a sequence profile, called PSSM. In
particular, we performed PSI-BLAST searches with
default parameters applying a threshold of E-value <
0.001 in the UniRef50 protein sequence database
(Suzek et al., 2015) as PSI-BLAST allows us to
discover protein sequences that are evolutionary
linked to the search sequence (Hamp and Rost, 2015;
Figure 1: Overall deep learning architecture to predict
interactions between viral and human host proteins.
Hashemifar et al., 2018). Sequence profiles for each
search sequence were processed by truncating
profiles of long sequences to a fixed length n and
zero-padding short sequences, a method widely used
for data pre-processing and effective training
(Matching, 2018; Min et al., 2017). As a result, we
obtained a 𝑛×20 dimensional array S for each
protein sequence, capturing the probability 𝑠
,
that
the residue in the i
th
position of the sequence is the j
th
out of the alphabet of 20 amino acids.
𝑆=
⎣
⎢
⎢
⎢
⎡
𝑠
,
⋯𝑠
,
⋯𝑠
,
⋮⋯⋮⋯⋮
𝑠
,
⋯𝑠
,
⋯𝑠
,
⋮⋯⋮⋯⋮
𝑠
,
⋯𝑠
,
⋯𝑠
,
⎦
⎥
⎥
⎥
⎤
,
2.1.2 Siamese CNN Module
To capture complex relationship between two
proteins we employ a Siamese CNN architecture with
two identical CNN sub-networks that share the same
parameters for a given pair of protein profiles 𝑆,𝑆
.
ICAART 2021 - 13th International Conference on Agents and Artificial Intelligence
42

Each sub-network produces a sequence embedding of
a single protein profile that are then concatenated.
While each single CNN module consists of a
convolutional and pooling layer, we leveraged four
connected convolutional modules to capture the
patterns in an input sequence profile.
Specifically, we use 𝑋, a 𝑛×𝑠 array of length 𝑛
with 𝑠 features in each position. The convolution
layer applies a sliding window of length 𝑤 (the size
of filters/kernels) to convert 𝑋 into a
(
𝑛−𝑤+1
)
×
𝑓𝑛 array 𝐶 where 𝑓𝑛 represents the number of
filters/kernels. Let 𝐶
,
denote the score of
filter/kernel 𝑘 , 1≤𝑘≤𝑓𝑛, that corresponds to
position 𝑖 of array 𝑋. Moreover, the convolutional
layer applies a parameter-sharing kernel 𝑀, a 𝑓𝑛 ×
𝑚 × 𝑠 array where 𝑀
,,
is the coefficient of pattern
𝑘 at position 𝑗 and feature 𝑙. The calculation of 𝐶 is
defined as
𝐶=𝐶𝑜𝑛𝑣
(
𝑆
)
𝐶
,
= 𝑀
,,
𝑋
,
Furthermore, the pooling layer is utilized to reduce
the dimension of 𝐶 to a
(
𝑛−𝑝+1
)
×𝑓𝑛 array 𝑃
where p is the size of pooling window. Array 𝑃=
𝑃𝑜𝑜𝑙
(
𝐶
)
is calculated as the maximum of all
positions 𝑖≤𝑗≤𝑖+𝑝 over each feature 𝑘 where
1≤𝑖≤
(
𝑛−𝑚+1
)
−𝑝,
𝑃
,
=𝑚𝑎𝑥𝐶
,
,…,𝐶
,
.
2.1.3 Prediction Module
The prediction module concatenates a pair of protein
sequence embedding vectors into a sequence pair
vector as the input of fully connected layers in an
MLP and computes the probability that two proteins
interact. The MLP contains three dense layers with
leaky ReLU where cross-entropy loss is optimized for
the binary classification objective defined as
𝐿𝑜𝑠𝑠 = −
1
|
𝐾
|
𝑦
𝑙𝑜𝑔
∈
𝑠
where 𝑦
is numerical class label of the protein pair 𝑝.
The output of the MLP for the protein pair 𝑝 is a
probability vector 𝑠̂
, whose dimensionality is the
number of classes 𝑚. s is normalized by a softmax
function, where the normalized probability value for
the 𝑖
class is defined as 𝑠
=
exp (𝑠̂
)
∑
exp (𝑠̂
)
.
2.1.4 Implementation Details
As for pre-acquired sequence profile construction, we
consider a fixed sequence length of 2,000. As for the
construction of our learning approach, we employ
four convolutional modules, with input size 20, 64,
128 and 256. The convolution kernel size is set to 3
while the size of pooling window is set to 2 with 3
max-pooling layers and a global max-pooling layer.
To optimize the cross-entropy loss function we use
AMSGrad (Reddi et al., 2018) and set the learning
rate to 0.0001. The batch size was set to 64, while the
number of epochs was 100. The fully connected
layers contain three dense layers with input size
1,024, 512, 256 and output a two-dimensional vector
with the last softmax layer. The whole procedure was
implemented with keras (https://keras.io/) with GPU
configuration.
2.2 Data Set Construction
We collected experimentally verified human-virus
PPI data capturing 9,880 interactions in HIV, 5,966
in Herpes, 5,099 in Papilloma, 3,044 in Influenza,
1,300 in Hepatitis, 927 in Dengue and 709 in Zika
from five public databases, including HPIDB
(Ammari et al., 2016), VirHostNet (Guirimand et al.,
2015), VirusMentha (Calderone et al., 2015),
PHISTO (Durmuş Tekir et al., 2013) and PDB
(Altunkaya et al., 2017). As for interactions of
proteins of SARS-CoV-2, we used two recently
published interaction sets (Gordon et al., 2020; Liang
et al., 2020) that captured 291 and 598 PPIs,
respectively. To obtain high-quality PPIs, we
removed interactions from large-scale mass
spectroscopy experiments that were detected only
once, non-physical interactions and interactions
between proteins without available PSSM features.
Sampling negative interactions, we applied our
‘Dissimilarity-Based Negative Sampling’ method as
outlined in our previous work (Yang et al., 2020).
Briefly, we sampled a negative training set of PPIs
(i.e. pairs of proteins that do not interact) by
considering interactions in the positive training set.
Given that we found a protein B with a sequence that
was similar to interacting protein A, we considered
B and C non-interacting. In particular, we sampled a
negative PPI set that was 10 times larger than the
positive PPI training set.
2.3 Transfer Learning
To further improve the performance of our deep
neural network especially when dealing with smaller
datasets, we propose two transfer learning methods
that keep the weights constant (i.e. frozen) or allow
Multi-scale Convolutional Neural Networks for the Prediction of Human-virus Protein Interactions
43

their fine-tuning in the early layers and applied them
to eight human-virus PPI sets. (i) We used the
proposed DNN architecture to train the models based
on a given source set of human-virus interactions to
obtain pre-trained weights in the CNN layers that
learn the representation of the protein sequences. (ii)
In subsequent transfer learning steps, we keep the
weights of these CNN layers constant (i.e. frozen) and
only re-train parameters of the fully connected layers
of the MLP to predict interactions in a target human-
viral interaction set. As an alternative, our fine-tuning
approach allows us to retrain the weights of CNN
layers that we obtained from the initial training step
and change such weights by learning the interactions
in a target set of human-virus interactions. In analogy
to the ‘frozen’ approach, we re-train parameters of the
fully connected layers of the MLP as well.
2.4 Alternative Machine Learning and
Feature Encoding Methods
A great amount of research demonstrates that
Random Forest (RF) algorithms perform better than
other machine learning methods when applied to
binary classification problems (Chen et al., 2019; Wu
et al., 2009; Yang et al., 2020). Therefore, we
compare the performance of our deep learning
approaches to this representative state-of-art
classifier. Moreover, we consider three widely-used
encoding methods for feature representations as the
input to the RF classifier.
2.4.1 Random Forest
Random Forest (F) (Hamp and Rost, 2015; Wu et al.,
2009) is an ensemble learning method where each
decision tree is constructed using a different bootstrap
sample of the data (‘bagging’). In addition, random
forests change how decision trees are constructed by
splitting each node, using the best among a subset of
predictors randomly chosen at that node (‘boosting’).
Compared to many other classifiers this strategy turns
out to be robust against over-fitting, capturing
aggregate effects between predictor variables. We
utilize the GridSearchCV function to optimize the
parameters for the RF algorithm and set the
‘neg_log_loss’ scoring function as the assessment
criterion.
2.4.2 Alternative Feature Encoding
Approaches
Amino acid sequences provide primary structure
information of a protein that work well as feature
representations of binary PPIs. Here, we use three
commonly used sequence-based encoding schemes
including Local Descriptor (LD) (Cui et al., 2007;
Davies et al., 2008; Tong and Tammi, 2008; Yang et
al., 2010), Conjoint Triad (CT) (Sun et al., 2017) and
Auto Covariance (AC) (Guo et al., 2008; You et al.,
2013). Generally, these features cover specific, yet
different aspects of protein sequences such as
physicochemical properties of amino acids,
frequency information of local patterns, and
positional distribution information of amino acids.
3 RESULTS AND DISCUSSION
3.1 Performance of the Proposed Deep
Learning Method
Applying our deep learning approach to a set of
different human-viral protein interaction data sets, we
observed generally high prediction performance of
our deep learning approach (Table 1). However, we
also found that small training data sets such as
Dengue, Zika and SARS-CoV-2 translated into
decreasing prediction performance.
Table 1: Performance of our deep learning architecture
(PSSM+CNN+MLP) using 5-fold cross validation.
Human-viral
PPI dataset
Sensitivity Specificity AUPRC
HIV
89.72 99.54 0.974
Herpes
68.10 97.98 0.768
Papilloma
70.48 98.53 0.818
Influenza
70.30 98.68 0.834
Hepatitis
49.77 97.79 0.636
Dengue
45.85 98.04 0.605
Zika
59.94 98.96 0.746
SARS-CoV-2
55.12 98.53 0.672
To compare the performance of our proposed
deep learning method (i.e. PSSM+CNN+MLP), we
trained a RF model using three widely used sequence-
based feature encoding schemes (i.e. LD, CT and AC)
on human-virus PPI datasets using 5-fold cross
validation. Comparing corresponding AUPRC
values, we observe that our method generally
outperformed other those RF based classifiers
especially when applied to comparatively large
datasets (Table 2). To further assess the impact of our
encoding scheme to represent the features of
interacting proteins, we compared the performance of
our deep learning architecture using PSSMs and a
different word embedding technique, word2vec+CT
one-hot. Specifically, this method considers each
amino acid as a word and learns a word-embedding
of sequences based on the training data, where each
amino acid is finally encoded by a 5-dimensional
ICAART 2021 - 13th International Conference on Agents and Artificial Intelligence
44

Table 2: Performance comparison of our deep learning
architecture (PSSM + CNN + MLP) and random forests
(RF) that were combined with three sequence encoding
schemes (LD, CT, AC) using 5-fold cross validation.
AUPRC
Human-
viral PPI
dataset
Our
method
LD+RF CT+RF AC+RF
HIV 0.974 0.972 0.97 0.972
Herpes 0.768 0.741 0.737 0.699
Papilloma 0.818 0.74 0.724 0.656
Influenza 0.834 0.813 0.795 0.713
Hepatitis 0.636 0.571 0.58 0.537
Dengue 0.605 0.526 0.505 0.456
Zika 0.746 0.720 0.718 0.698
SARS-
CoV-2
0.672 0.668 0.678 0.652
vector. Moreover, the 20 amino acids can be clustered
into 7 groups based on their dipoles, volumes of the
side chains and other chemical descriptors.
Furthermore, CT one-hot is a 7-dimensional one-hot
encoding based on the classification of these 20
amino acids. As a result, word2vec+CT one hot is the
concatenation of pre-trained word embeddings and
CT one-hot encodings for each protein that is
represented by a 𝑛×13 dimensional array. As noted
previously, we considered a fixed sequence length of
n = 2,000 and zero-padded smaller sequences. In
comparison to word2vec+CT one hot, Table 3
indicates that our learning approach combined with
PSSM allows better prediction performance
especially in comparatively small datasets such as
Dengue, Zika and SARS-CoV-2.
3.2 Comparison with Several Existing
Human-virus PPI Prediction
Methods
To further assess the performance of our proposed
method, we compared our method with three existing
human-virus PPI prediction approaches. Recently, we
proposed a sequence embedding-based RF method to
predict human-virus PPIs with comparatively
promising performance (Yang et al., 2020). The main
point of our approach is the application of an
unsupervised sequence embedding technique (i.e.
doc2vec) to represent protein sequences as low-
dimensional vectors with rich features. Such
representations of protein pairs were subjected to a
RF method that predicted the presence/absence of an
interaction. In Alguwaizani et al.’s work
(Alguwaizani et al., 2018), the authors utilized a
Support Vector Machine (SVM) model to
predict human-virus PPIs based on a simple way to
feature-encode protein sequences through repeat
patterns and local patterns of amino acid
combinations. As for the DeNovo method (Eid et al.,
2016), the authors introduced a domain/linear motif-
based SVM approach to predict human-virus PPIs. To
compare, we first constructed the PSSMs of the
Table 3: Performance comparison of combinations of
different feature encodings (PSSM, word2vec+CT one-hot)
and our deep learning architecture (CNN + MLP).
AUPRC
Human-viral
PPI dataset
PSSM
word2vec+
CT one hot
HIV 0.974 0.968
Herpes 0.768 0.734
Papilloma 0.818 0.778
Influenza 0.834 0.808
Hepatitis 0.636 0.587
Dengue 0.605 0.481
Zika 0.746 0.662
SARS-CoV-2 0.672 0.602
protein sequences of DeNovo’s PPI dataset to train
our learning model. Finally, we assessed the
performance of our reconstructed deep learning
model on the test set provided in (Eid et al., 2016)
including 425 positive and 425 negative samples.
Table 4: Performance comparison of our method (PSSM +
CNN+MLP) with existing human-virus PPI prediction
methods.
Method
Accuracy
(%)
Sensitivity
(%)
Specificity
(%)
Our model 94.12 90.82 97.41
doc2vec+RF
a
93.23 90.33 96.17
SVM
b
86.47 86.35 86.59
DeNovo
c
81.90 80.71 83.06
a
The corresponding values were retrieved from (Yang et
al., 2020).
b
The corresponding values were retrieved from
(Alguwaizani et al., 2018).
c
The corresponding values were
retrieved from (Eid et al., 2016).
Furthermore, we tested our previous RF based
prediction method and Alguwaizani et al’s SVM
approach on these data sets as well. Table 4 clearly
suggests that our deep learning and previously
published RF based method outperformed
Alguwaizani et al.’s SVM and the DeNovo approach.
Multi-scale Convolutional Neural Networks for the Prediction of Human-virus Protein Interactions
45
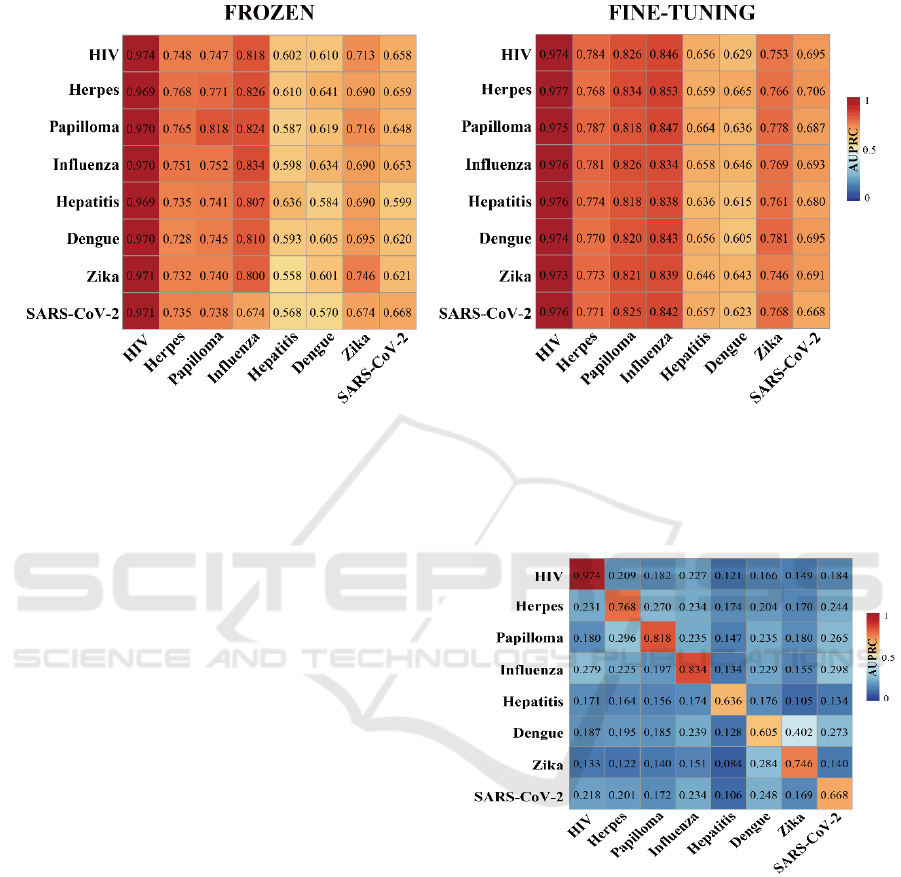
Table 4: Performance comparison of our method (PSSM + CNN+MLP) with existing human-virus PPI prediction methods.
3.3 Cross-viral Tests and Transfer
Learning
To explore potential factors that affect prediction
performance in a cross-viral setting, we trained our
deep learning model on one human-virus PPI data set
and predicted protein interactions in a different
human-virus system. Expectedly, such cross-viral
tests dropped considerably in performance compared
to training and testing in the same human-viral system
(Fig. 2). To allow reliable cross-viral predictions of
PPIs, we introduce two transfer learning methods
where we trained the parameters of CNN layers of the
DNN model on a source human-virus PPI dataset.
Subsequently, we transfer all parameters to initialize
a new model (i.e. frozen or fine tuning) to train on a
target human-virus PPI dataset. To comprehensively
test our transfer learning approaches, we considered
each combination of human-viral PPI sets as source
and target data. The left panel in Fig. 3 indicates that
a relatively rigid transfer learning methodology by
keeping the parameters of the feature encoding CNN
untouched (i.e. frozen) strongly outperformed
baseline performance as shown in Fig. 2. In turn, fine-
tuning parameters using a given target human-viral
domain allowed for another marked increase in
performance (right panel, Fig. 3) compared to the
‘frozen’ approach. As for individual pairs of human-
viral domains, we also observed that the frozen
transfer methodology worked well if the target
domain data set was large, independently of the
training domain. In turn, performance dropped when
the target human-viral domain datasets of PPIs were
small. Notably, prediction performance improved
when we applied our fine-tuning transfer learning
approach on small target domains data sets such as
human-Hepatitis, human-Dengue, human-Zika and
human-SARS-CoV-2.
Figure 2: AUPRC performance of cross-viral tests. Rows
indicate human-viral PPIs that were used for training while
columns indicate human-viral PPI test sets.
4 CONCLUSIONS
Here, we proposed a Siamese-based multi-scale CNN
architecture by using PSSM to represent the
sequences of interacting proteins, allowing us to
predict interactions between human and viral proteins
with an MLP approach. In comparison, we observed
that our model outperformed previous state-of-the-art
human-virus PPI prediction methods. Furthermore,
ICAART 2021 - 13th International Conference on Agents and Artificial Intelligence
46

we confirmed that the performance of the
combination of our deep learning framework and the
representation of the protein features as PSSMs was
mostly superior to combinations of other machine
learning and pre-trained feature embeddings. While
we found that our model that was trained on a given
source human-viral interaction data set performed
dismally in predicting protein interactions of proteins
in a target human-virus domain, we introduced two
transfer learning methods (i.e. frozen type and fine-
tuning type). Notably, our methods increased the
cross-viral prediction performance dramatically,
compared to the naïve baseline model. In particular,
for small target datasets, fine-tuning pre-trained
parameters that were obtained from larger source sets
increased prediction performance.
REFERENCES
Alguwaizani, S., Park, B., Zhou, X., Huang, D.S., and Han,
K. (2018). Predicting interactions between virus and
host proteins using repeat patterns and composition of
amino acids. J. Healthc. Eng. 2018, 1391265.
Altunkaya, A., Bi, C., Bradley, A.R., Rose, P.W., Prli, A.,
Christie, H., Costanzo, L. Di, Duarte, J.M., Dutta, S.,
Feng, Z., et al. (2017). The RCSB protein data bank:
integrative view of protein, gene and 3D structural
information. Nucleic Acids Res. 45, D271–D281.
Ammari, M.G., Gresham, C.R., McCarthy, F.M., and
Nanduri, B. (2016). HPIDB 2.0: a curated database for
host-pathogen interactions. Database 2016, baw103.
Calderone, A., Licata, L., and Cesareni, G. (2015).
VirusMentha: a new resource for virus-host protein
interactions. Nucleic Acids Res. 43, D588–D592.
Chang, H., Han, J., Zhong, C., Snijders, A.M., and Jian-
Hua, M. (2018). Unsupervised transfer learning via
multi-scale convolutional sparse coding for biomedical
applications. IEEE Trans. Pattern Anal. Mach. Intell.
40, 1182–1194.
Chen, M., Ju, C.J.T., Zhou, G., Chen, X., Zhang, T., Chang,
K.W., Zaniolo, C., and Wang, W. (2019). Multifaceted
protein-protein interaction prediction based on Siamese
residual RCNN. Bioinformatics 35, i305–i314.
Cheplygina, V., de Bruijne, M., and Pluim, J.P.W. (2019).
Not-so-supervised: a survey of semi-supervised, multi-
instance, and transfer learning in medical image
analysis. Med. Image Anal. 54, 280–296.
Cui, J., Han, L.Y., Li, H., Ung, C.Y., Tang, Z.Q., Zheng,
C.J., Cao, Z.W., and Chen, Y.Z. (2007). Computer
prediction of allergen proteins from sequence-derived
protein structural and physicochemical properties. Mol.
Immunol. 44, 514–520.
Davies, M.N., Secker, A., Freitas, A.A., Clark, E., Timmis,
J., and Flower, D.R. (2008). Optimizing amino acid
groupings for GPCR classification. Bioinformatics 24,
1980–1986.
Durmuş Tekir, S., Çakir, T., Ardiç, E., Sayilirbaş, A.S.,
Konuk, G., Konuk, M., Sariyer, H., Uǧurlu, A.,
Karadeniz, I., Özgür, A., et al. (2013). PHISTO:
pathogen-host interaction search tool. Bioinformatics
29, 1357–1358.
Eid, F., Elhefnawi, M., and Heath, L.S. (2016). DeNovo:
virus-host sequence-based protein-protein interaction
prediction. 32, 1144–1150.
Gordon, D.E., Jang, G.M., Bouhaddou, M., Xu, J.,
Obernier, K., White, K.M., O’Meara, M.J., Rezelj, V.
V., Guo, J.Z., Swaney, D.L., et al. (2020). A SARS-
CoV-2 protein interaction map reveals targets for drug
repurposing. Nature 583, 459–468.
Guirimand, T., Delmotte, S., and Navratil, V. (2015).
VirHostNet 2.0: surfing on the web of virus/host
molecular interactions data. Nucleic Acids Res. 43,
D583–D587.
Guo, Y., Yu, L., Wen, Z., and Li, M. (2008). Using support
vector machine combined with auto covariance to
predict protein-protein interactions from protein
sequences. Nucleic Acids Res. 36, 3025–3030.
Hamp, T., and Rost, B. (2015). Evolutionary profiles
improve protein-protein interaction prediction from
sequence. 31, 1945–1950.
Hashemifar, S., Neyshabur, B., Khan, A.A., and Xu, J.
(2018). Predicting protein-protein interactions through
sequence-based deep learning. Bioinformatics 34,
i802–i810.
Karimi, M., Wu, D., Wang, Z., and Shen, Y. (2019).
DeepAffinity: interpretable deep learning of
compound-protein affinity through unified recurrent
and convolutional neural networks. Bioinformatics 35,
3329–3338.
Kriegeskorte, N., and Douglas, P.K. (2018). Cognitive
computational neuroscience. Nat. Neurosci. 21, 1148–
1160.
Le, Q. V., Karpenko, A., Ngiam, J., and Ng, A.Y. (2011).
ICA with reconstruction cost for efficient overcomplete
feature learning. Adv. Neural Inf. Process. Syst. 24 25th
Annu. Conf. Neural Inf. Process. Syst. 2011, NIPS
2011 2027–2035.
Lee, H., Grosse, R., Ranganath, R., and Ng, A.Y. (2009).
Convolutional deep belief networks for scalable
unsupervised learning of hierarchical representations.
Proc. 26th Annu. Int. Conf. Mach. Learn. ICML 54,
609–616.
Liang, Q., Li, J., Guo, M., Tian, X., Liu, C., Wang, X.,
Yang, X., Wu, P., Xiao, Z., Qu, Y., et al. (2020). Virus-
host interactome and proteomic survey of PMBCs from
COVID-19 patients reveal potential virulence factors
influencing SARS-CoV-2 pathogenesis. BioRxiv
2020.03.31.019216.
Matching, S. (2018). Neural article pair modeling for
wikipedia sub-article matching. In: ECML-PKDD 3–
19.
Min, X., Zeng, W., Chen, N., and Chen, T. (2017).
Chromatin accessibility prediction via convolutional
long short-term memory networks with k -mer
embedding. Bioinformatics 33, i92–i101.
Multi-scale Convolutional Neural Networks for the Prediction of Human-virus Protein Interactions
47

Pospisil, D.A., Pasupathy, A., and Bair, W. (2018).
’Artiphysiology’ reveals V4-like shape tuning in a deep
network trained for image classification. Elife 7,
e38242.
Reddi, S.J., Kale, S., and Kumar, S. (2018). On the
convergence of Adam and Beyond. 6th Int. Conf.
Learn. Represent. ICLR 2018 - Conf. Track Proc. 1–23.
Sainath, T.N., Kingsbury, B., Saon, G., Soltau, H.,
Mohamed, A. rahman, Dahl, G., and Ramabhadran, B.
(2015). Deep convolutional neural networks for large-
scale speech tasks. Neural Networks 64, 39–48.
Shao, L., Zhu, F., and Li, X. (2015). Transfer learning for
visual categorization: a survey. IEEE Trans. Neural
Networks Learn. Syst. 26, 1019–1034.
Sun, T., Zhou, B., Lai, L., and Pei, J. (2017). Sequence-
based prediction of protein protein interaction using a
deep-learning algorithm. BMC Bioinformatics 18, 277.
Suzek, B.E., Wang, Y., Huang, H., McGarvey, P.B., and
Wu, C.H. (2015). UniRef clusters: a comprehensive and
scalable alternative for improving sequence similarity
searches. Bioinformatics 31, 926–932.
Taroni, J.N., Grayson, P.C., Hu, Q., Eddy, S., Kretzler, M.,
Merkel, A., Greene, C.S., Therapeutics, T., Diseases,
S., Arbor, A., et al. (2019). MultiPLIER: a transfer
learning framework for transcriptomics reveals
systemic features of rare disease Jaclyn. 8, 380–394.
Tong, J.C., and Tammi, M.T. (2008). Prediction of protein
allergenicity using local description of amino acid
sequence. 13, 6072–6078.
Wu, J., Liu, H., Duan, X., Ding, Y., Wu, H., Bai, Y., and
Sun, X. (2009). Prediction of DNA-binding residues in
proteins from amino acid sequences using a random
forest model with a hybrid feature. Bioinformatics 25,
30–35.
Yamins, D.L.K., and DiCarlo, J.J. (2016). Using goal-
driven deep learning models to understand sensory
cortex. Nat. Neurosci. 19, 356–365.
Yang, L., Xia, J.-F., and Gui, J. (2010). Prediction of
protein-protein interactions from protein sequence
using local descriptors. Protein Pept. Lett. 17, 1085–
1090.
Yang, X., Yang, S., Li, Q., Wuchty, S., and Zhang, Z.
(2020). Prediction of human-virus protein-protein
interactions through a sequence embedding-based
machine learning method. Comput. Struct. Biotechnol.
J. 18, 153–161.
You, Z.-H., Li, L., Ji, Z., Li, M., and Guo, S. (2013).
Prediction of protein-protein interactions from amino
acid sequences with ensemble extreme learning
machines and principal component analysis. BMC
Bioinformatics 14, 80–85.
Zhang, S., Zhou, J., Hu, H., Gong, H., Chen, L., Cheng, C.,
and Zeng, J. (2016). A deep learning framework for
modeling structural features of RNA-binding protein
targets. Nucleic Acids Res. 44, e32.
ICAART 2021 - 13th International Conference on Agents and Artificial Intelligence
48
