
The Trap of 2D in Artificial Models of Tumours: The Case for 3D
In-silico Simulations
Dario Panada
a
and Bijan Parsia
b
Department of Computer Science, The University of Manchester, Oxford Road, Manchester, U.K.
Keywords:
Agent-based Models, In-silico Modelling, Bio-oncology, Cancer, Simulations.
Abstract:
Artificial modelling of tumours can provide insights in cancer biology and offer a powerful complement to
laboratory research. A common approach is to simulate tumour growth in a two-dimensional environment
and to then generalize results to a three-dimensional one. Literature suggests this strategy fails to adequately
capture the underlying biology and may provide misleading results. To establish whether 2D models may form
a viable alternative to 3D ones, we developed a model comprising cancer cell growth and proliferation and
soluble diffusion to replicate experiments previously performed in a laboratory. We made use of established
parametrization techniques to configure our simulations and novel error estimation strategies to evaluate them.
Our results suggest that the same simulation in 2D and 3D yields significantly different results. Further, that the
cause of this discrepancy lies in the spatial geometry of 2D simulations which does not allow for the formation
of hypoxic regions in the tumour mass. We conclude with a recommendation that due to the limitations of
2D simulations, and the negligible difference in cost between the two approaches, 3D simulations should be
employed over 2D ones.
1 INTRODUCTION
Artificial modelling of tumour development is a grow-
ing discipline in the field of oncology. Widespread
availability of powerful computer clusters and cloud
computing resources mean that scenarios comprising
large sections of tissue and high number of cells can
be easily simulated in reduced amounts of time. The
results of these investigations can be used to inform
research and promote either further iterations of the
in-silico process or in-vivo or in-vitro laboratory ex-
periments.
In-silico simulations offer several advantages as
precursors or complements to laboratory wetware re-
search. They allow to rapidly explore multiple ’what-
if’ scenarios, simulating weeks if not months of tu-
mour growth in the space of hours. They further al-
low a high resolution of measurements and observa-
tions, with properties of individual cells being observ-
able. And finally, they allow to investigate ’causa-
tion vs correlation’ problems in instances where mul-
tiple phenomena could be responsible for a behavior
of the tumour mass. This latter is particularly impor-
tant when devising new therapies, as it is essential that
a
https://orcid.org/0000-0001-9004-5592
b
https://orcid.org/0000-0002-3222-7571
the target phenomenon is actually the one driving the
behavior we wish to suppress.
The two most common approaches to cancer mod-
elling are agent-based models and continuous sim-
ulations. In the former, agents of different classes
are used to represent different cells or group of cells.
Each agent class is assigned a set of rules that governs
its evolution and interaction with the environment and
other cells. In the latter, partial differential equations
are used to describe changes in concentration of cells
at different positions. These are then solved to ob-
serve the evolution of the tumour mass and adjacent
tissues in time.
Both approaches require a process of spatial dis-
cretization, where the section of tissue being inves-
tigated is fit to a structure such as a grid or mesh.
For agent-based models, this is so that each position
may be occupied by one or more agents. Whereas
for continuous systems the solution to the equations
will determine the concentration of cells at each posi-
tion. The most simple example of such discretization
involves a Cartesian coordinate system, where the ge-
ometry of the grid is consistent across the space and
each position’s volume or area is constant. More ad-
vanced techniques involve meshes with positions of
different sizes or shapes, which allows for varying
Panada, D. and Parsia, B.
The Trap of 2D in Artificial Models of Tumours: The Case for 3D In-silico Simulations.
DOI: 10.5220/0010517702390247
In Proceedings of the 11th International Conference on Simulation and Modeling Methodologies, Technologies and Applications (SIMULTECH 2021), pages 239-247
ISBN: 978-989-758-528-9
Copyright
c
2021 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
239

levels of resolution across the environment.
A decision that has to be made early in the de-
sign process is whether the spatial discretization will
produce a two dimensional (2D) or a three dimen-
sional (3D) environment. The case for 2D being
that it closely resembles cancer growth in a Petri
dish, a commonly adopted approach in laboratories,
whereas 3D would allow to more closely mimic tu-
mour growth in living tissues.
It has been suggested that advantages of 3D mod-
elling over 2D include better capturing of oncogene
activation (Pickl and Ries, 2009), protein expres-
sion and drug sensitivity (Melissaridou et al., 2019;
Imamura et al., 2015; Lv et al., 2017) as well as
more realistic biochemical and biomechanical envi-
ronments (Duval et al., 2017) and a better translation
of pathophysiological features of the tumour environ-
ment (Hoarau-V
´
echot et al., 2018). Finally, 3D mod-
els have also been suggested to better capture inter-
cellular signalling pathways. (Riedl et al., 2017)
An initial survey of the literature reveals a con-
siderable number of publications relying on 2D dis-
cretization strategies to obtain insights in tumour bi-
ology. These include, among others, studies of blood
vessel development in response to angiogenic stimuli
(Gabhann et al., ), response to chemotherapy (Sinek
et al., 2004) and tumour oxygenation (Skeldon et al.,
2012). Additional searches for publications in the
field of tumour modelling revealed a large corpus of
simulations using a 2D environment. (Zhang et al.,
2011; Turner and Sherratt, 2002; Sun et al., 2012; An-
derson and Chaplain, 1998; Cai et al., 2011) In cases
where 3D was chosen an approach (Olsen and Siegel-
mann, 2013; Shirinifard et al., 2009; Cai et al., 2016),
it is not explained why this was decided for rather than
opting for a 2D approach.
Previous related work (St et al., 2005) highlighted
differences in simulation performance between 2D
and 3D, but did not make an assessment regarding the
suitability or unsuitability of either.
Many studies using 2D models do not address the
implications and potential limitations of modelling
in 2D over 3D. Effects on the spatial distribution of
agents, evolution of the tumour mass and soluble dif-
fusion are not addressed, nor is there any attempt,
practical of theoretical, at mapping results back to a
3D environment. Finally, it is often not clear how de-
cisions regarding the effective size of a grid position
or number of biological cells represented by an agent
were taken. While these are model artifacts and prop-
erties of the simulation, changes in values could affect
the model’s output and support or invalidate a thesis.
With these items not addressed and the wealth of
literature advocating 3D over 2D, and a substantial
corpus of literature opting for both 2D and 3D ap-
proaches, we wish to investigate whether it is the case
that 2D may offer a cheaper and accurate alternative
to 3D or if simulations should on the other hand be
preferably or exclusively performed in three dimen-
sions.
The rest of the paper is structured as follows. In
our Materials and methods section we present our
model setup, parametrization strategy and our ap-
proach to mapping values from a 3D to a 2D envi-
ronment. In our Results section we present our ex-
perimental findings for the various simulations in 2D
and 3D, alongside measurements regarding tumour
properties such as oxygenation levels, etc. Finally,
in the Discussion we draw a conclusion with regards
to whether 2D form an adequate approach to in-silico
investigations or if 3D should be preferably or neces-
sarily employed.
We implemented a simple agent-based model
with continuous elements to include diffusion of sol-
ubles. Key phenomena accounted for include cellular
growth and proliferation, blood vessel development in
response to secretion of vascular endothelial growth
factors (VEGF) and hypoxia and spatial heterogene-
ity with regards to oxygen concentration.
We believe that if, despite the simplicity of our
model, issues in the 3D to 2D translation still mani-
fest then these would also be present in more complex
models. Adding more biology would therefore only
make these worse. In summary, the simplicity of our
model allows us to explore the effects of 3D to 2D
mapping of simulations while minimizing additional
work needed to implement more complex behaviours
and decide for values in models with a higher amount
of parameters.
Our initial setup in 3D was derived from starting
conditions reported in medical literature. A trans-
lation to 2D was then derived from this, and sim-
ulations were run in 3D and 2D. Finally, resulting
growth curves were compared to those reported in
medical publications to determine whether any dif-
ference in error rates were significant. Where pos-
sible, parameter values were obtained from literature.
Where this was not possible, either because specific
values or unknown or because contrasting values are
published, hyper-parameter tuning techniques such as
grid search (Rios et al., 2013) and random search
(Bergstra JAMESBERGSTRA and Yoshua Bengio
YOSHUABENGIO, 2012) were employed to explore
a suitable search-space.
SIMULTECH 2021 - 11th International Conference on Simulation and Modeling Methodologies, Technologies and Applications
240

1.1 Model Overview
We now provide a high-level overview of our model,
including key dynamics and properties of agents.
1.1.1 Spatial Discretization
Space is discretized as a Cartesian system. Each posi-
tion in the grid has its own concentration of solubles
(Eg: oxygen) and can host a certain amount of agents.
In the 2D model coordinates are identified by pairs of
values (Eg: (2,4)), in 3D by triplets (Eg: (2,4,6)).
1.1.2 Temporal Discretization
Time is advanced in discrete steps of two hour. This
corresponds to the length of the shortest stage of the
cell life-cycle: Mitosis. At each epoch, soluble con-
centrations at each position are updated and so is the
state of each agent. While in theory each agent is up-
dated simultaneously, this is practically not possible
and an order of execution needs to be specified. To
avoid systematic bias due to certain agents being al-
ways updated first, the scheduling order is random-
ized at the start of each epoch.
1.1.3 Cell Life-cycle
Our model includes two classes of cells: Cancer cells
and endothelial cells. The latter will be discussed in
the section dedicated to angiogenesis, whereas here
we will be explaining the former.
The main concept behind cancer cells is that of
uncontrolled proliferation. Given a sufficient oxygen
concentration, cancer cells will keep growing and di-
viding until a physical constraint owing to their sur-
rounding being saturated occurs. To account for var-
ious factors that may delay the division of individual
cells, a probability value is set for cells to transition
from Growth I (G1) into Synthesis. Upon complet-
ing G1, a cancer cell transitions into Synthesis with
a given probability which is set as a simulation pa-
rameter. If the cell does not transition into Synthesis,
it will attempt to do so at the following epoch with
the same probability until it does. Once a cell pro-
gresses into Synthesis, it will proceed to completing
the cell life-cycle and then divide into two daughter
cells. The only exception would be if its surrounding
environment is saturated, where then it would not be
able to further divide.
A cancer cell may be Active, Quiescent or Dead.
Active cells are growing and proliferating, quiescent
cells are not progressing in the cell life-cycle and are
secreting VEGF and dead cells simply contribute to
the tumour’s volume but no longer have any active
role. Two oxygen concentration thresholds are speci-
fied as simulation parameters: O
Hypoxia
and O
Critical
,
which determine the state of a cancer cell. The re-
lation between oxygen concentration at a given po-
sition O and the state of a cell is detailed in equa-
tion 1. The transition from active to quiescent is re-
versible if oxygen concentrations subsequently rise
again above O
Hypoxia
. Logically, a dead cell may how-
ever not transition back into other states. We assume
O
Hypoxia
> O
Critical
> 0.
state =
Active, if O ≥ O
Hypoxia
,
Quiescent, if O ≥ O
Critical
,
Dead otherwise
; (1)
1.1.4 Diffusion of Solubles
Our model incorporates the diffusion of oxygen from
endothelial cells to cancer cells and of VEGF from
cancer cells to endothelial cells. Concentrations of
each soluble are calculated at the start of each epoch
with values for each environment position updated.
Given a soluble κ, equation 2 governs the diffusion
process.
∂κ
∂t
= D
κ
∇
2
+ s
κ
− u
κ
(2)
D
κ
is the diffusion coefficient, s
κ
the source rate and
u
κ
the uptake (or sink) rate. The equations are solved
using FiPy (Guyer et al., 1988) which implements the
finite-volume method and we setup our solution using
no-flux boundary conditions.
Diffusion of solubles from the intracellular space
to the extracellular matrix occurs by diffusion, which
means the concentration outside cells may not be
greater than that inside cells. As such, source rates of
individual cells may be adjusted and reduced to avoid
implausible scenarios such as soluble flow against a
concentration gradient. Where the inner and outer
concentrations are equal the source rate effectively
becomes zero.
Individual sink rates may also be adjusted to avoid
negative substrate concentrations, although a mini-
mum uptake rate must be maintained owing for ex-
amples to cells needing at least some level of oxygen.
If even then a negative concentration is obtained, this
indicates that the number of cells at a position exceeds
the capacity of the blood vessel network and a num-
ber of cells equal to the amount required to restore
positive concentrations is considered dead.
1.1.5 Angiogenesis
Angiogenesis refers to the development of new blood
vessels from existing ones in response to VEGF
The Trap of 2D in Artificial Models of Tumours: The Case for 3D In-silico Simulations
241
stimuli. Tumours secrete VEGF in conditions of
hypoxia to increase the oxygen and nutrient supply.
In our model, we differentiate endothelial cells in Tip
and Trunk cells.
Trunk cells are static agents. They act as sources
of oxygen but do not proliferate or otherwise interact
with the environment. Tip cells, on the other hand,
exhibit a more dynamic behavior. Given a minimum
VEGF concentration, set as a model parameter, Tip
cells will migrate up-gradient to an adjacent grid posi-
tion. The position originally occupied will host a new
Trunk cell. In-between elongations, Tip cells must
complete one full cell life-cycle.
1.2 Experimental Setup
We now proceed to illustrating our experimental
setup, our derivation of 2D environments from 3D
ones and parametrization techniques.
1.2.1 The Target Curve
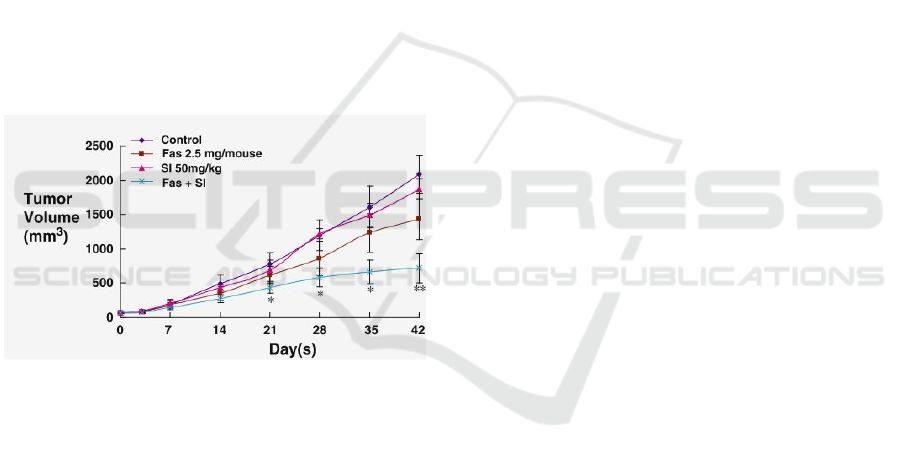
Figure 1: The target growth curves. We will be bench-
marking our model against the ”control” curve over the first
25 days.
We will be bench-marking against the control curve
reported by Chen et al. (Chen et al., ) and shown
in Figure 1, comparing the tumour volume obtained
in our simulation to that obtained in empirical stud-
ies. We will be considering the first 2 weeks of tu-
mour growth so as to keep the problem computation-
ally tractable and allow us to repeat large amounts
of simulations on our infrastructure. In picking the
publication we would use as reference, we privileged
those which reported clearly readable growth curves
with a sufficient number of data-points and smaller
error bars.
1.2.2 Spatial Discretization and
Transformations
As mentioned earlier, the environment is discretized
as a Cartesian grid. Table 1 summarizes the effective
sizes of grid positions, alongside other relevant spatial
values such as the volume of cancer cells and initial
tumour volume. Derived 2D values are also reported.
1.2.3 Model Resolution
A decision that needs to be made upfront is the reso-
lution of the model. That is to say, how many cancer
cells will be represented by an agent. This is a com-
promise between model reliability and computational
cost. A high number of cancer cells per agent will
mean overall fewer agents necessary, but lowers the
resolution (Ie: We capture a lower degree of cellu-
lar heterogeneity.), whereas a low number of cancer
cells per agent provides a higher resolution but higher
computational costs. Different resolutions also af-
fect the maximum number of agents per grid position
and the required initial number of agents to achieved
the desired start volume or area. We will be testing
three resolutions: 300, 600 and 1,200 cancer cells per
agent. These are summarized in table 2.
Testing a resolution of fewer than 300 cancer cells
per agent becomes computationally intractable, and
decreasing the resolution to exceed 1,200 cancer cells
per agent results in 2D simulations having a maxi-
mum number of agents per position of 1, which hin-
ders either tumour growth or blood vessel develop-
ment.
1.2.4 Starting Conditions
Cancer cells in an amount appropriate for a given res-
olution are seeded at the center of the grid. Every po-
sition in the grid is seeded with one endothelial cell.
Initial soluble concentrations are calculated and as-
signed to positions. The simulation is thereafter al-
lowed to run its course.
2 RESULTS
Each simulation set (Eg: 300 cancer cells per agent,
2D) comprised 200 simulations. These shared the
same parameters assigned as constant values but dif-
fered in values for those assigned by grid search or
random search and for those related to spatial quanti-
ties such the as maximum number of agents per posi-
tion. Each simulation produced a growth curve which
was compared to an expected growth curve derived
SIMULTECH 2021 - 11th International Conference on Simulation and Modeling Methodologies, Technologies and Applications
242

Table 1: Summary of spatial units for 2D and 3D simulations. 2D values were derived by assuming 3D structures were
cuboids, and then deriving the area of the base. Values are shown to two decimal places.
3D 2D
Item Value Item Value
Volume of Cancer Cell 2,000.00 µm
3
Area of Cancer Cell 158.74 µm
2
Volume of Grid Position 0.30 mm
3
Area of Grid Position 0.45 mm
2
Start Tumour Volume 1.20 mm
3
Start Tumour Area 1.13 mm
2
Table 2: Summary of how different model resolutions affect the maximum number of agents that can be held in a grid position
and the number of agents required at the start to obtain the desired initial volume or area. As expected, increasing the number
of cancer cells per agent decreases both the carrying capacity of individual positions and the initial number of required agents.
Cancer Cells per Agent Max Agents per
Grid Position
Initial Number of
Agents
300
2D 9 24
3D 500 2,000
600
2D 4 12
3D 250 1,000
1,200
2D 2 6
3D 125 500
from medical literature. This gave an indication of the
error rate for such simulation. A set’s performance is
calculated as the average error rate of all simulations
in it. This allows, for each resolution, to compare the
performance of 2D and 3D simulations. For 2D sim-
ulations, an extra step is needed to map areas back to
volumes.
2.1 The Error Function
The aim of the error function is to compare the tu-
mour growth curve produced by a simulation (in-
silico curve) to the one observed in empirical lab-
oratory studies (empirical curve). And ultimately,
to produce a single value: The absolute mean error.
This provides an indication of how closely an in-silico
growth curve aligns to the empirical growth curve.
Expected tumour volumes at different time-points
were estimated from the empirical growth curve (Fig-
ure 1).
A polynomial was then fit to these points describing
the expected volume (V
exp
) at a day (d). This is re-
ported as equation 5, with coefficients reported to two
decimal places.
V
exp
(d) = 0.95d
2
+ 18.10d − 65.68; 0 ≤ d ≤ 25 (3)
Given the actual volume at a day d reported in a sim-
ulation’s growth curve, V
d
act
, then the error ε
d
is as
reported in equation 4 the magnitude of the difference
between the expected and actual value.
ε
d
= |V
d
act
−V
exp
(d)| (4)
The absolute mean error of a simulation (ε) is then
calculated as the average of all errors calculate at each
time-point:
ε = avg(ε
0
, ε
1
, ..., ε
d−1
, ε
d
);0 ≤ d ≤ 25 (5)
2.2 Estimating Volumes from Areas
For 2D simulations, these obviously returned tumour
areas. In order to compare these to expected volumes,
the area was assumed to be the base of a 3D struc-
ture and the corresponding volume was derived via a
simple mathematical transformation.
The generic form of this basic transformation is
provided in equation 6, where V indicates volume and
a area. It represents the derivation of a cube’s volume
from a square’s area by obtaining the square’s width
and raising it to the cube.
V (a) = a
3
2
(6)
2.3 Resulting Error Rates
Error rates for 2D and 3D simulations across all res-
olutions are show in figure 2. For higher resolutions
(Ie: Lower numbers of cancer cells represented by a
single agent), 3D clearly outperforms 2D. At lower
resolutions (Ie: Higher numbers of cancer cells rep-
resented by a single agent) it is the case that the two
approaches appear to be comparable or that 2D out-
performs 3D. This is in fact misleading and will be
addressed in our discussion.
The Trap of 2D in Artificial Models of Tumours: The Case for 3D In-silico Simulations
243

We note the stability of the error in 3D, where
discrepancies of ±150mm
3
are owed to the stochas-
tic nature of the model. As for 2D, we note that the
error seems to be linearly dependent and negatively
correlated to the resolution. While it might appear 2D
could be as good as 3D in some instances, this is not
the case and is further elaborated in our discussion.
Finally, a Welch’s t-test was performed compar-
ing the populations of errors for 2D vs. 3D at each
resolution. Results were significant for across all res-
olutions for p < 10
−10
. This t-test is appropriate as it
does not assume equal population variance.
3 DISCUSSION
Our results suggest a relatively small and stable er-
ror when it comes to 3D simulations, independently
of the resolution. More interesting is the 2D case,
which seems to outperform 3D at lower resolutions
and whose error seems to increase linearly as resolu-
tion is increased.
This might initially be interpreted as 2D outper-
forming 3D at all but the highest resolutions, suggest-
ing that the former offers a viable if not preferable
alternative to the latter. In fact, such is not the case.
The apparent better performance of 2D is an artifact
of the specific model configuration which coinciden-
tally allows it to minimize the absolute mean error.
We will discuss how 2D simulations suffer a funda-
mental flaw due to dimensionality mapping from 3D.
We will also emphasize the importance of validation
strategies which go beyond growth curve comparison,
but also consider elements such as soluble concentra-
tions and phenotype ratios within the cancer cell pop-
ulations. Without these additional verifications our
analysis would be incomplete and lead to the conclu-
sion that 2D forms an alternative to 3D, whereas this
is not the case.
In the first instance we note the direction of the
mean error for 2D simulations. Errors are calculated
as the difference between actual and expected vol-
umes. So, a positive error means the model predicted
a larger volume than expected, and a negative one
that the model predicted a smaller volume than ex-
pected. A summary is reported in table 3 (see ap-
pendix). For resolutions of 300 and 600 cancer cells
per agent all simulations consistently over-estimate
tumour growth. For a resolution of 1,200 cancer
cells per agent, a decisive majority of simulation over-
estimates tumour growth. It therefore appears fair to
state that 2D models systematically over-estimate tu-
mour growth.
Given that the magnitude of the error of 2D simula-
tions increases as resolution increases, and that 2D
systematically over-estimate growth, we can say that
as resolution increases 2D simulations over-estimate
growth by larger amounts.
To explain this we need to consider several factors.
First, that as we increase resolution we decrease the
number of cells per agent and therefore we increase
the initial number of agents needed to keep the start-
ing volume consistent. Given more initial agents it
makes sense that the final tumour volume should be
greater and that therefore the error should increase,
but this does not explain why we observe this in 2D
but not in 3D.
The reason behind this lies in the effect that 3D
to 2D mapping and resolution adjustments combined
have on the maximum number of agents per position.
In 2D, a position may host between 2 and 9 agents. In
3D, between 125 and 500. (See tables 1 and 2.) This
means that in 3D we may obtain a much higher cancer
to endothelial ratio at each position than in 2D. This
is because cancer cells produce daughter cells each
capable of dividing, resulting in exponential growth,
whereas a tip cell will only produce another tip cell
and trunk cell, the latter being unable to divide, lead-
ing to linear growth.
In both 3D and 2D cancer cells will exceed en-
dothelial cells but, crucially, in 2D space constraints
means the ratio will never increase to the point where
oxygen becomes a limiting factor. This is owed to
the source rate of oxygen being much higher than its
sink rate, and an endothelial cell being able to support
multiple cancer cells. Unless enough space is allowed
at a position for a significant amount of cancer cell
divisions, these will never exceed the capacity of the
initial blood vessel network.
In summary, in 2D simulations oxygen never be-
comes a limiting factor of growth meaning that a
higher number of initial cancer cells, as is seeded at
higher resolutions, results in a larger final tumour vol-
ume. In 3D instead oxygen may become a limiting
factor and modulates and restricts tumour growth.
Our results raise important considerations for tu-
mour modelling. Where simulations implement a 2D
environment, appropriate scales and parameter values
need to be chosen to avoid issues similar to those we
described which would make the models biologically
implausible. Specifically, parameter values derived
from 3D studies (such as empirical ones in labora-
tories) should not simply be copied over to 2D in-
silico experiments without consideration being given
to the change of dimensionality. However, publica-
tions rarely discuss the process of deriving their 2D
environments from 3D ones.
Consideration should be given to the use of re-
SIMULTECH 2021 - 11th International Conference on Simulation and Modeling Methodologies, Technologies and Applications
244

Figure 2: Summary of error rates for each resolution, 2D vs. 3D. Error bars represent standard deviation on the mean. A
higher number of cancer cells per agent indicates a lower resolution, as this means the simulation is modelling larger groups
of cells together rather than allowing them to develop independently.
Table 3: For each resolution we report the number of 2D simulations which over-estimate tumour growth (positive error)
against the number that under-estimate tumour growth (negative error).
Resolution (Cancer
Cells per Agent)
Number of Positive
Errors
Number of Negative
Errors
300 196 0
600 194 0
1,200 187 12
medial action to make 2D models viable. For exam-
ple, by implementing strategies to increase sink rates
or decrease source rates of oxygen. Or, alternatively
and among other possible approaches, decoupling the
maximum number of cells per position from the over-
lying 3D model from which a 2D one has supposedly
been derived. While these approaches may produce
apparently valid results, the question then is what im-
pact does the aforementioned set of remedial actions
have on the reliability of results. For example, setting
the maximum number of agents per cell to an arbi-
trary value may result in a model accepting unrealis-
tic cell concentrations in tissues. Or, tweaking source
and sink values may result in the model working with
hyper- or under-oxygenated tissues.
More importantly, enforcing artificial constraints
on the model subtracts from its emergent behaviors.
The main value of these models lies in them revealing
unexpected and insightful patterns from a set of sim-
ple, underlying rules. Developers should specify the
behavior of individual agents so that they mimic as
closely as possible their biological counterparts. De-
velopers should not try to direct the evolution of the
model in a specific direction as they would be doing
when imposing remedial strategies. To do so would
be to constrain the simulation setting it up towards a
specific conclusion or end-state, which would limit its
usefulness towards gaining additional understanding
of biological phenomena.
It is worth adding some biological considerations
to the limitations of 2D models we discussed. Along-
side unrestricted growth where oxygen is no longer a
limiting factor, other properties of tumours will re-
sult as altered. In 2D we will not observe regions
of hypoxia in the cancer mass, nor the formation of
a necrotic core. Secretion of VEGF will not occur,
which in turn will lead to no angiogenesis or blood
vessel developent. In simulations where these were
implemented, hypoxia-inducible factor (HIF) path-
ways would not be activated. HIF pathways medi-
ate significant phenotypic alterations in cancer cells
(Philip et al., 2013; Semenza, 2007), which would
The Trap of 2D in Artificial Models of Tumours: The Case for 3D In-silico Simulations
245

therefore not be observed in 2D. It is therefore clear
how the impact of oxygen concentrations on 2D mod-
els significant affects the accuracy and reliability in a
negative manner.
We also emphasize the numerical scale of our sim-
ulations. Confidence in our results is further rein-
forced by running 200 simulations per simulation set.
(A simulation set refers to, for example, the group
of 2D simulations having 300 cancer cells per agent)
This reduces the likelihood of results being due the
stochastic nature of the model, allowing us to infer
that they are truly representative of the model’s be-
havior.
This contrasts with other publications surveyed,
where the number of simulations run is not discussed.
Given models often include elements of stochastic-
ity, it is important for multiple iterations of these to
be evaluated so as to obtain a clearer picture of their
underlying behavior and evolution. Multiple runs
also allow to explore the search space for parameters
whose value has not been established with certainty,
assessing the model’s sensitivity to changes in these
and informing us about the impact on the results of
the simulation. Search strategies for parametrization,
such as but not limited to grid and random search,
should be explicitly mentioned and discussed. Distri-
bution parameters should be reported, as well as the
number of draws from each distribution and relevant
information. This is necessary for experimental re-
producibility.
We will now consider costs. We ran our sim-
ulations on Amazon AWS EC2 instances of class
r4.large. These feature 2 vCPUs and 16GiB of RAM.
The cost of these is between $0.01 and $0.001 per
hour depending on the specific plan selected. A sin-
gle 2D simulation took on average one hour and a sin-
gle 3D simulation on average four hours. We will also
note that while we used a large fleet of instances to run
a high number of simulations, individual simulations
could be run in reasonable time within an average of-
fice laptop.
Hence, the price of simulations in 2D or 3D is
comparable. Even for the more expensive 3D simu-
lations, a single simulation set comprising 200 simu-
lations will still cost under $1. Given the severe limi-
tations of 2D we discussed, and the negligible differ-
ences in computational and dollar cost, we think our
investigation makes a very strong case for 3D as the
sole viable solution.
In summary, careless or approximate mapping of
3D environments into 2D ones, or creation of 2D envi-
ronments without consideration for their 3D counter-
parts, raises questions about the validity of the model.
Where this discussion is omitted, as is the case in
many publications, this impacts the reliability of re-
sults as it is not clear what accuracy or resolution has
been achieved by the model. Further, due to the neg-
ligible difference in costs between 2D and 3D, we be-
lieve there is a clear case for the latter to be preferred
over the former.
4 CONCLUSION
We have developed an agent-based model of tumour
growth with continuous elements to account for solu-
ble diffusion and simulated tumour growth in 3D and
derived 2D environments at multiple resolutions of
cancer cells per agent. We have addressed the issue
of parametrization by wherever possible relying on
values in literature, and where these were unknown
using well-established search techniques such as grid
search and random search.
Error rates for each simulation were calculated by
comparing growth curves produced by our model to
those reported in literature, and at each resolution we
compared error distributions and average error val-
ues for 2D and 3D. Initial results suggested, purely
on based on error rates, that 3D simulations consis-
tently produced low error rates and outperformed 2D
at higher resolutions. On the other hand, 2D simula-
tions outperformed 3D at lower resolutions and their
error was dependent on the resolution.
Further analysis revealed that the combined im-
pact of cancer cells per agent resolution and 3D to 2D
mapping significantly altered the topology of the 2D
environment. Because of this oxygen concentrations,
a factor limiting growth in 3D, no longer affected tu-
mour growth in 2D. As a result, tumour growth in 2D
became predominantly a function of the initial vol-
ume and was no longer modulated by oxygen avail-
ability as is the case in 3D.
These results highlight the challenges of simulat-
ing a phenomenon that occurs in 3D, such as tumour
growth, in 2D. Neglecting to explicitly derive the 2D
environment from a 3D one, as many publications do,
raises questions about the validity of the model and
realibility of the results.
In conclusion, our results suggest that 3D mod-
els should be preferred to 2D ones. In cases where
3D ones may not be implemented, a clear discussion
should be provided regarding the derivation of the 2D
model from a 3D one.
SIMULTECH 2021 - 11th International Conference on Simulation and Modeling Methodologies, Technologies and Applications
246

REFERENCES
Anderson, a. R. and Chaplain, M. a. (1998). Contin-
uous and discrete mathematical models of tumor-
induced angiogenesis. Bulletin of mathematical biol-
ogy, 60(5):857–899.
Bergstra JAMESBERGSTRA, J. and Yoshua Bengio
YOSHUABENGIO, U. (2012). Random Search for
Hyper-Parameter Optimization. Journal of Machine
Learning Research, 13:281–305.
Cai, Y., Wu, J., Li, Z., and Long, Q. (2016). Mathematical
Modelling of a brain tumour initiation and early de-
velopment: A coupled model of glioblastoma growth,
pre-existing vessel co-option, angiogenesis and blood
perfusion. PLoS ONE, 11(3):1–28.
Cai, Y., Xu, S., Wu, J., and Long, Q. (2011). Coupled
modelling of tumour angiogenesis, tumour growth
and blood perfusion. Journal of Theoretical Biology,
279(1):90–101.
Chen, Y., Alvarez, E. A., Azzam, D., Wander, S. A., Gug-
gisberg, N., Jord
`
a, M., Ju, Z., Hennessy, B. T., and
Slingerland, J. M. Combined Src and ER blockade
impairs human breast cancer proliferation in vitro and
in vivo.
Duval, K., Grover, H., Han, L. H., Mou, Y., Pegoraro, A. F.,
Fredberg, J., and Chen, Z. (2017). Modeling physio-
logical events in 2D vs. 3D cell culture.
Gabhann, F. M., Ji, J. W., and Popel, A. S. Computational
Model of Vascular Endothelial Growth Factor Spatial
Distribution in Muscle and Pro-Angiogenic Cell Ther-
apy.
Guyer, J. E., Wheeler, D., and Warren, J. A. (1988). FiPy:
Partial Differential Equations with Python. Comput-
ing in Science Engineering, 11(3):6–15.
Hoarau-V
´
echot, J., Rafii, A., Touboul, C., and Pasquier,
J. (2018). Halfway between 2D and animal mod-
els: Are 3D cultures the ideal tool to study cancer-
microenvironment interactions?
Imamura, Y., Mukohara, T., Shimono, Y., Funakoshi, Y.,
Chayahara, N., Toyoda, M., Kiyota, N., Takao, S.,
Kono, S., Nakatsura, T., and Minami, H. (2015).
Comparison of 2D- and 3D-culture models as drug-
testing platforms in breast cancer. Oncology Reports,
33(4):1837–1843.
Lv, D., Hu, Z., Lu, L., Lu, H., and Xu, X. (2017). Three-
dimensional cell culture: A powerful tool in tumor re-
search and drug discovery.
Melissaridou, S., Wiechec, E., Magan, M., Jain, M. V.,
Chung, M. K., Farnebo, L., and Roberg, K. (2019).
The effect of 2D and 3D cell cultures on treatment
response, EMT profile and stem cell features in head
and neck cancer 11 Medical and Health Sciences 1112
Oncology and Carcinogenesis. Cancer Cell Interna-
tional, 19(1).
Olsen, M. M. and Siegelmann, H. T. (2013). Multiscale
agent-based model of tumor angiogenesis. Procedia
Computer Science, 18:1016–1025.
Philip, B., Ito, K., Moreno-S
´
anchez, R., and Ralph, S. J.
(2013). HiF expression and the role of hypoxic mi-
croenvironments within primary tumours as protective
sites driving cancer stem cell renewal and metastatic
progression. Carcinogenesis, 34(8):1699–1707.
Pickl, M. and Ries, C. H. (2009). Comparison of 3D
and 2D tumor models reveals enhanced HER2 acti-
vation in 3D associated with an increased response to
trastuzumab. Oncogene, 28(3):461–468.
Riedl, A., Schlederer, M., Pudelko, K., Stadler, M., Wal-
ter, S., Unterleuthner, D., Unger, C., Kramer, N.,
Hengstschl
¨
ager, M., Kenner, L., Pfeiffer, D., Krupitza,
G., and Dolznig, H. (2017). Comparison of cancer
cells in 2D vs 3D culture reveals differences in AKT-
mTOR-S6K signaling and drug responses. Journal of
Cell Science, 130(1):203–218.
Rios, L. M., Nikolaos, ·., Sahinidis, V., and Rios, L. M.
(2013). Derivative-free optimization: a review of
algorithms and comparison of software implementa-
tions. J Glob Optim, 56:1247–1293.
Semenza, G. L. (2007). HIF-1 mediates the Warburg effect
in clear cell renal carcinoma. Journal of Bioenergetics
and Biomembranes, 39(3):231–234.
Shirinifard, A., Gens, J. S., Zaitlen, B. L., Popławski, N. J.,
Swat, M., and Glazier, J. A. (2009). 3D multi-cell
simulation of tumor growth and angiogenesis. PLoS
ONE, 4(10).
Sinek, J., Frieboes, H., Zheng, X., and Cristini, V. (2004).
Two-dimensional chemotherapy simulations demon-
strate fundamental transport and tumor response limi-
tations involving nanoparticles. Biomedical Microde-
vices, 6(4):297–309.
Skeldon, A. C., Chaffey, G., Lloyd, D. J., Mohan, V.,
Bradley, D. A., and Nisbet, A. (2012). Modelling
and detecting tumour oxygenation levels. PLoS ONE,
7(6):1–29.
St, A., ˜phanou, ., Mcdougall, S. R., Anderson, A. A. A.,
and Chaplain, M. A. J. (2005). MATHEMATI-
CAL AND Mathematical Modelling of Flow in 2D
and 3D Vascular Networks: Applications to Anti-
Angiogenic and Chemotherapeutic Drug Strategies.
COMPUTER MODELLING Mathematical and Com-
puter Modelling, 41:1137–1156.
Sun, X., Zhang, L., Tan, H., Bao, J., Strouthos, C., and
Zhou, X. (2012). Multi-scale agent-based brain cancer
modeling and prediction of TKI treatment response:
Incorporating EGFR signaling pathway and angiogen-
esis. Technical report.
Turner, S. and Sherratt, J. A. (2002). Intercellular adhesion
and cancer invasion: A discrete simulation using the
extended potts model. Journal of Theoretical Biology,
216(1):85–100.
Zhang, L., Jiang, B., Wu, Y., Strouthos, C., Zhe Sun, P.,
Su, J., and Zhou, X. (2011). Developing a multi-
scale, multi-resolution agent-based brain tumor model
by graphics processing units. Technical report.
The Trap of 2D in Artificial Models of Tumours: The Case for 3D In-silico Simulations
247
