
Molecular Fragments from Incomplete, Real-life NMR Data:
Framework for Spectra Analysis with Constraint Solvers
Haneen A. Alharbi
1
a
, Igor Barsukov
2 b
, Rudi Grosman
2 c
and Alexei Lisitsa
1 d
1
Department of Computer Science, University of Liverpool, Liverpool, U.K.
2
Institute of Systems, Molecular and Integrative Biology, University of Liverpool, Liverpool, U.K.
Keywords:
Constraint Satisfaction Problem, Constraint Programming, NMR Data Interpretation, Molecular Structure
Generation.
Abstract:
Nuclear Magnetic Resonance (NMR) spectroscopy is a powerful analytical tool that can be used in the eluci-
dation of chemical structures and is widely applied both in academia and industry. Despite using computer-
assisted structure elucidation systems, interpretation of NMR data is often laborious, requires high levels of
expertise and is not immune to ambiguities. In this multi-disciplinary study, we developed a design of a novel
system using a Constraint Satisfaction (CS) framework to utilise unannotated NMR spectra. Additionally,
our system allows the utilisation of complementary information obtained/known outside the scope of NMR.
Herein we describe a prototype implementation and its empirical evaluation on a set of amino acids, which are
a diverse class of important biological compounds. We further employ the CS approach to show the principle
limits (ambiguity) of the NMR method in molecular structure elucidation.
1 INTRODUCTION
Nuclear Magnetic Resonance (NMR) spectroscopy
is a cornerstone analytical method widely used both
in academia and industry. One of the main appli-
cations of this method is chemical structure eluci-
dation (Elyashberg and Williams, 2015). There are
different types of NMR techniques to collect par-
tial structural information on molecular structures of
substances under investigation. An example of the
type of structural information which can be obtained
from NMR spectra is that a molecule should con-
tain a Carbon (C) atom directly bonded to a Hydro-
gen (H) atom. Various other types of structural in-
formation are available from the spectra (Elyashberg
and Williams, 2015) which makes it possible to iden-
tify/elucidate the structure of the molecule, which
constitute the main goal of NMR analysis. The elu-
cidation process is complex, laborious and the re-
sults may be ambiguous, so the interpretationof NMR
spectra currently requires the involvement of human
experts. The research on the automation of the NMR
a
https://orcid.org/0000-0002-0281-8346
b
https://orcid.org/0000-0003-4406-9803
c
https://orcid.org/0000-0002-0233-7112
d
https://orcid.org/0000-0002-3820-643X
spectra interpretation and development of Computer-
Assisted Structure Elucidation (CASE) systems has
been conducted since the 1960s (Koichi et al., 2014)
and several such systems are available either as re-
search prototypes or commercial propositions (Burns
et al., 2019).
There are still many remaining challenges in
computer-assisted NMR analysis: full automation (or
greater degree of automation), managing the uncer-
tainty of spectra, handling the ambiguity of the anal-
ysis or dealing with the mixtures as opposed to pure
substances. The main task of NMR analysis lends it-
self very naturally to the CS area. Indeed, the par-
tial structural information obtained from NMR spec-
tra can be seen as a set of constraints, with an eluci-
dated molecular structure being a solution to this set
of constraints. This observation was a starting point
of our investigation and we found it quite surprising
that no attempts to apply generic CS techniques (as
opposed to specialised algorithms) to NMR analysis
have been made until very recently. In (Omrani and
Naanaa, 2016; Omrani and Naanaa, 2019) the authors
have demonstrated that the basic tasks of NMR analy-
sis can be solved by reformulation of the structural in-
formation obtained from NMR spectra as constraints
and the application of generic constraint solvers. The
open-source system (Omrani and Naanaa, 2019) al-
834
Alharbi, H., Barsukov, I., Grosman, R. and Lisitsa, A.
Molecular Fragments from Incomplete, Real-life NMR Data: Framework for Spectra Analysis with Constraint Solvers.
DOI: 10.5220/0010915800003116
In Proceedings of the 14th International Conference on Agents and Artificial Intelligence (ICAART 2022) - Volume 3, pages 834-841
ISBN: 978-989-758-547-0; ISSN: 2184-433X
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

lows applying basic types of NMR constraints as well
as user-specified allowed/forbidden molecular struc-
tures. While this work has demonstrated the viability
of the CS-based NMR analysis, important theoretical
and practical questions have been left open. First, the
power of the proposed method has been illustrated by
the case studies but was not systematically assessed.
Second, the NMR constraints were considered from
the idealised perspective, and no practical limitations
of the NMR method were taken into account. For
the latter, in the practice of NMR analysis, it is quite
common that some theoretically possible NMR con-
straints are not extractable from the spectra. Further-
more, the ambiguity of the NMR structure elucidation
was not addressed.
In this paper, we present the design of the system
for NMR interpretation/molecular structure elucida-
tion based on the CS. We take as a principle an inher-
ent incompleteness and uncertainty of NMR derivable
partial information about the molecular structures.
We consider several classes of constraints, including
chemical constraints, such as valency of atoms, NMR
constraints such as chemical bond connectivity and
other constraints coming either from other types of
analysis (e.g. mass spectrometry) or from the prac-
tical experience. We discuss the formulation of vari-
ous types of constraints and report on the experiments
with a prototype implementation on the classes of
amino acids. We also show that NMR analysis is in-
herently ambiguous, even when all theoretically pos-
sible NMR constraints are available, there are cases
when it is still impossible to uniquely identify the
molecular structure in question. Thus, we assess the
fundamental limitation of the NMR method itself.
2 PRELIMINARIES
We will represent the molecular structures as labelled
undirected multigraphs, which are formally defined
as triples hV, e, li, where V is a set of vertices, e :
V × V → Z
≥
represents the multiplicity of edges be-
tween the vertices, and l : V → A labels the vertices
by the types of chemical elements. Here A is a set of
all chemical elements, including e.g.
H
for
hydrogen
,
C
for
carbon
, etc. We use multigraphs as opposed
to graphs to represent faithfully the cases of molec-
ular structures with multiple bonds between pairs of
atoms. A function v : A → Z
+
denotes the valency,
fundamental chemical characteristic of the atom types
that denotes the maximum capacity of making bonds.
For example v(H) = 1 and v(C) = 4.
We will deal in this paper with two main types of
2-D (two-dimensional) NMR spectra used in the elu-
cidation of molecular structures: Heteronuclear Sin-
gle Quantum Coherence (HSQC) and Heteronuclear
Multiple Bond Correlation (HMBC) experiments that
correlate signals of
13
C and
1
H atoms. Typically these
spectra are visualised as contour plots where the axes
coordinates are called chemical shifts with the units
of Hertz or the most commonly used normalised scale
parts per million (ppm) by convention.
A molecule’s structural information appears in the
spectra in the form of peaks shown as the small spots
at the intersection of the chemical shifts of the inter-
acting atoms (see Figure 1 (left) and (right) for peaks
in the HSQC and HMBC spectra, respectively). Each
peak in the HSQC spectrum corresponds to (or is gen-
erated by) a pair of
C
and
H
atoms with a direct bond
between them, meaning the graph-distance between
corresponding vertices in the representing multigraph
is 1. Similarly, the peaks in the HMBC spectrum iden-
tify the pairs of
C
and
H
atoms separated by two or
three bonds (i.e distance between corresponding ver-
tices in the multigraph is 2 or 3).
For both types of spectra, x-coordinate, and y-
coordinate of a peak represent chemical shifts of cor-
responding
H
and
C
atoms, respectively. The chemical
shifts of the atoms persist across different spectra and
different atoms may have the same (very close) chem-
ical shifts due to possible symmetries of the molecule.
Figure 1 illustrates these principles, where the molec-
ular structure in the middle produces HSQC (left) and
HMBC (right) spectra. The (in)equalities of chemi-
cal shifts are subject to possible measurement errors
and subtle differences in an experimental setup, and
they should be considered as approximate. We as-
sume here, as a starting point in constraints formu-
lation, that all necessary approximations/abstractions
have been done and it is firmly established which co-
ordinates (shifts) are equal and which are different.
This is sufficient to formulate the basic NMR con-
straints (see Section 3.3). Furthermore, the informa-
tion about the shifts taking values in a specific range
is useful for NMR interpretation and to formulate fur-
ther NMR constraints (see Section 3.4).
The problem of NMR analysis/interpretation we
consider here can be reformulated as Given a set of
NMR-derivable constraints from HSQC and HMBC
spectra on the connections between atoms to gener-
ate all molecular multigraphs satisfying those con-
straints. Additional structural information derived
from other NMR experiments can be introduced by
extending the constraint set. The ultimate goal of
such an analysis is to identify the molecular structures
down to the least ambiguous set possible. In the next
section, we discuss the important aspects of constraint
formulation for this problem.
Molecular Fragments from Incomplete, Real-life NMR Data: Framework for Spectra Analysis with Constraint Solvers
835

Figure 1: The connectivity information from HSQC (left panel blue arrow) and HMBC (right panel red arrows) obtained for
alanine (middle panel). Additional NMR information such as chemical shifts can be added as optional constraints and are
planned to be implemented in the future. NMR Spectra are courtesy of James London.
3 CONSTRAINTS FOR NMR
ANALYSIS
In the constraint-based NMR analysis, several cat-
egories of constraints do naturally occur. Some of
them are absolute/rigid, in a sense, they have to be
present and satisfied in all reasonable scenarios of
analysis, while others are soft constraints that may
be present/absent and are not always required to be
satisfied. The latter categories reflect inherent incom-
pleteness, uncertainty and practical limitations of the
NMR method. Further to that, we consider two dif-
ferent settings for the analysis, in one we seek com-
plete solutions, meaning the full molecular structures,
in another, we seek incomplete molecular structures
or fragments. The incomplete setting is important, as
NMR-derivable information is often insufficient to re-
cover the full molecular structure and other sources
of information are needed, but the recovered frag-
ments may be still useful for NMR spectra interpre-
tation. Yet another aspect of NMR interpretation is
whether an analysis of pure substances or mixtures is
required. All these variants of the problem affect the
ways some constraints are formulated. In this sec-
tion we discuss the constraints in an implementation-
independent form; the details of the implementation
of a prototype system can be found in Section 4.
3.1 Molecular Graph Representation
Constraints
The solutions of NMR CS are thought in terms of la-
belled undirected multigraphs, so the following rigid
constraints are necessary:
• e(x, y) ≥ 0 (M)ultigraph;
• e(x, y) = e(y, x) (U)ndirected;
• e(x, x) = 0 (N)o Loops;
Due to the additivity of NMR spectra when ap-
plied to mixtures as opposed to pure substances, NMR
derived constraints are related to all participating
molecules. In the case of the analysis of pure sub-
stances as opposed to the mixtures, the solutions need
to represent a single molecule, so the connectedness
constraint is required:
• the molecular multigraph is connected
(C)onnectedness
Notice that to consider the case of mixtures with
unknown numbers of components it is sufficient just
to omit C constraint - the disconnected multigraphs
solutions will then include representations of the
molecular structures of the components. Due to the
complexity of the connectedness constraint, we pro-
pose to treat it at the post-processing stage, that is not
to include it as a constraint, generate all solutions and
then filter them on connectedness condition.
3.2 Basic Chemistry Constraints
All molecular structures are constrained by the va-
lency, the fundamental chemical property of the con-
stituting atoms. This imposes the following con-
straints depending on the settings:
• ∀x ∈ V d(x) = v(l(x)) (complete setting) (VC)
• ∀x ∈ V d(x) ≤ v(l(x)) (incomplete setting) (VI)
Here d(x) is a degree of a vertex x in a multigraph,
that is Σ
y∈V
e(x, y), v is a (predefined) valency func-
tion, and l is a atom type labelling function.
3.3 NMR Constraints
It is significant to express the HSQC and HMBC spec-
tra clearly in terms of constraints. The HSQC exper-
ICAART 2022 - 14th International Conference on Agents and Artificial Intelligence
836

iment identifies a single bond between the pairs of
C
and
H
atoms and the HMBC spectroscopy detects cor-
related atoms separated by two or three bonds in the
multigraphs. Let the HSQC and HMBC spectra con-
tain the peaks p
i, j
with 1 ≤ i ≤ t
1
;1 ≤ j ≤ t
2
and q
k,m
with 1 ≤ k ≤ s
1
;1 ≤ m ≤ s
2
, respectively. Let the co-
ordinates (chemical shifts) of these peaks be (h
i
, c
j
)
and (h
k
, c
m
), respectively. We will use the following
useful abbreviations:
dist
(1)
(x, y) for e(x, y) > 0 (The HSQC peaks),
dist
(2)
(x, y) for ∃z (e(x,z) > 0 ∧ e(z, y) > 0),
and dist
(3)
(x, y) for ∃z, v (e(x, z) > 0 ∧ e(z, v) > 0 ∧
e(v, y) > 0) (The HMBC peaks).
Then basic NMR constraints are defined as:
• ∃ ¯x∃ ¯y (HSQC( ¯x, ¯y) ∧ HMBC( ¯x, ¯y) ∧ ID( ¯x, ¯y))
(NMR)
where:
• ¯x = x
1
, . . . x
m
and ¯y = y
1
, . . . y
n
are sequences of
variables
• HSQC( ¯x, ¯y) is
V
i, j
(dist
(1)
(x
h
i
, y
c
j
) ∧ l(x
h
i
) = H ∧
l(y
c
j
) = C)
• HMBC( ¯x, ¯y) is
V
k,m
((dist
(2)
(x
h
k
, y
c
m
) ∨
dist
(3)
(x
h
k
, y
c
m
)) ∧ l(x
h
k
) = H ∧ l(y
c
m
) = C)
• ID( ¯x, ¯y) is
V
c
i
6=c
j
(x
c
i
6= x
c
j
) ∧
V
h
i
6=h
j
(y
h
i
6= y
h
j
)
Thus, the basic NMR constraints assert the ex-
istence of pairs of
C
-
H
atoms satisfying necessary
distance conditions, imposed by HSQC and HMBC
spectra (HSQC( ¯x, ¯y), HMBC( ¯x, ¯y), respectively), as
well as by identity conditions (ID( ¯x, ¯y)). Notice that
in the case when some peak coordinates are equal
(within one spectrum or across both), e.g. c
i
= c
j
then
corresponding variables y
c
i
, y
c
j
are the same.
3.4 Further Constraints
3.4.1 Exact/Partial Formula Constraints
Due to the partiality of NMR-derived information,
any additional information can be very useful for
structure elucidation. Other forms of analysis, in-
cluding different variants of spectroscopy (e.g. Mass-
Spectroscopy (MS)), may provide partial or full in-
formation on a Molecular Formula (MF), that is the
count of different types of atoms involved in the
molecule. Thus, for any known count n of a type of
atom T involved in a molecule adds a formula con-
straint:
• |{x | l(x) = T}| = n (F)
Figure 2: The workflow of the system. Demonstrating how
the system works for different data inputs.
3.4.2 Optional Constraints
It is not uncommon to have prior information on ex-
pected or impossible fragments during structure elu-
cidation and use it as optional constraints. This infor-
mation can be derived either from the knowledge of
the work being done or from the NMR data itself. For
example, if the analysed mixture is obtained through
a chemical synthesis all the precursor materials used,
the by-products and the products are known. If no
cyclic structures (e.g phenyl rings) are present, this
information can be imposed as an optional constraint
to prohibit cyclic solutions. Similarly, any expected
fragments can also be forced to the solution space.
3.5 Post-processing
In the proposed design, we assume that some of the
constraints may be computationally expensive to deal
with by a generic constraint solving or depending on
the solver, may be infeasible to express in its input
language. In such cases, the constraints can be used
for filtering the solutions at the post-processing stage.
In the implemented prototype we experimented with
the connectedness constraint being used for filter-
ing at the post-processing stage. Furthermore, post-
processing can be used to further reduce the number
of solutions by removing equivalent multigraphs. In
this vein, we experimented with partial/full isomor-
phism checks and filtering of obtained multigraphs.
Molecular Fragments from Incomplete, Real-life NMR Data: Framework for Spectra Analysis with Constraint Solvers
837

4 PROPOSED FRAMEWORK
AND PROTOTYPE
IMPLEMENTATION
The proposed high-level design of the constraint-
based NMR interpretation system from the discussed
principles is presented in Figure 2. The system sup-
ports different sources of the input data - it may come
either as pre-processed experimental spectra (lists of
peaks/chemical shifts) or as ideal constraints gener-
ated from known molecular structures data (molecu-
lar files). The latter is used for the testing and the
investigation of inherent ambiguity related to the CS
approach for NMR data analysis. Once all constraints
are formulated, based on the input data, a generic
constraint solver is applied which results in the set
of all possible molecular structures (labelled multi-
graphs) satisfying the constraints. Post-processing
(e.g. connectedness, or isomorphism filtering) is ap-
plied after that, and if the results remain ambigu-
ous/inconclusive, the set of constraints can be updated
and processing repeated.
The current prototype system is implemented in
Python and constraints programming platform Num-
berjack (Hebrard et al., 2010), and its constraint
solver Mistral (Hebrard, 2008) is used. The open-
source RDKit software (Landrum, 2016) is used
to handle the connection between atoms and draw
molecular structures. To identify isomorphic struc-
tures and to enumerate all nonisomorphic ones, we
used a standalone Nauty tool, which relies on canoni-
cal labelling algorithms (McKay and Piperno, 2014).
In this work we have used Nauty to check isomor-
phism of labelled graphs.
The performance of the system has been tested
by using known structures of chemical compounds.
Therefore, the NMR data for simple amino acids are
chosen from the Biological Magnetic Resonance Data
Bank (BMRB) website to be the data set for the sys-
tem (Ulrich et al., 2007).
5 EXPERIMENTS, RESULTS AND
EVALUATION
All the experiments were implemented on Intel Core
CPU’s with frequency 2.20 GHz running Ubuntu
18.04.5 and using 7 GB of RAM.
To test our approach we generated full sets of
HSQC and HMBC constraints for all 20 amino acids
from their chemical structures and calculated incom-
plete solutions and part of complete solutions (Ta-
ble 1 and 2, respectively). In each case, the solu-
tions were subsequently filtered at the post-processing
stage to find all connected and nonisomorphic multi-
graphs. In Table 1,
C
and
H
atoms were considered
in the incomplete setting experiment. For 18 out of
20 amino acids, we reported the number of all possi-
ble solutions with time in seconds and the number of
nonisomorphic structures with the time taken to fil-
ter the solutions in seconds. For two amino acids, we
stated an ’out of memory’ case while running the sys-
tem. To tackle this case, we might need further op-
timisation for the current implementation, including
using different constraint solvers. Multiple solutions
were generated by the constraint solver for amino
acids, with the number of solutions increasing signif-
icantly with the increased complexity of the molec-
ular structure, represented by the number of atoms
in a Molecular Formula (MF). Much higher number
of solutions was generated when the MF was used as
additional source of constraints (complete solutions),
compared to the incomplete solution set. Isomorphic
filtering only partially reduced the number of solu-
tions, demonstrating intrinsic ambiguity of the con-
straint sets.
Inspection of the partial solutions for alanine in
Figure 3 reveals several reasons for multiple solu-
tions. Most of the solutions are linear structures
where the
C
atom that does not have
H
atom attached
(no corresponding HSQC peak) is positioned either
at the end of the chain or between the two atoms
that have
H
atoms. This type of ambiguity is caused
by the intrinsic ambiguity of HMBC constraints that
correspond to either two or three-bond separation be-
tween the interacting atoms. This ambiguity can be
resolved by introducing additional NMR connectivity
constraints.
The second source of ambiguity is uncertainty in
the number of
H
atoms attached to each
C
. Since
C
has
Table 1: Incomplete solutions calculated for amino acids
using only NMR data as the input.
Amino acid MF Structures
# Sols Time (s) Nonisomorphic Time (s)
Alanine C
3
H
7
NO
2
25 0.00 15 0.00
Arginine C
6
H
14
N
4
O
2
1952 0.32 716 0.00
Asparagine C
4
H
8
N
2
O
3
104 0.00 60 0.00
Aspartic acid C
4
H
7
NO
4
104 0.00 60 0.00
Cysteine C
3
H
7
NO
2
S 25 0.00 15 0.00
Glutamine C
5
H
10
N
2
O
3
456 0.04 172 0.00
Glutamic acid C
5
H
9
NO
4
456 0.04 172 0.00
Glycine C
2
H
5
NO
2
3 0.00 3 0.00
Histidine C
6
H
9
N
3
O
2
6204 0.81 2060 0.02
Isoleucine C
6
H
13
NO
2
1977 0.33 427 0.01
Leucine C
6
H
13
NO
2
2279 0.33 346 0.01
Lysine C
6
H
14
N
2
O
2
2555 0.42 676 0.01
Methionine C
5
H
11
NO
2
S 910 0.11 163 0.00
Phenylalanine C
9
H
11
NO
2
136221 35.85 30735 0.69
Proline C
5
H
9
NO
2
603 0.12 85 0.00
Serine C
3
H
7
NO
3
25 0.00 15 0.00
Threonine C
4
H
9
NO
3
142 0.01 39 0.00
Tryptophan C
11
H
12
N
2
O
2
out of memory - - -
Tyrosine C
9
H
11
NO
3
out of memory - - -
Valine C
5
H
11
NO
2
603 0.14 85 0.00
ICAART 2022 - 14th International Conference on Agents and Artificial Intelligence
838

a valency of four, a terminal
C
can have up to three
H
atoms attached, and
C
in the middle of the linear
chain can have maximum two
H
atoms. The num-
ber of
H
atoms can be determined in the multiplicity-
edited HSQC experiment. The number of possible
variants caused by these two ambiguities increase dra-
matically with the number of
C
atoms in the molecule,
as clearly seen from Table 1 and 2.
The third type of ambiguity is caused by the pos-
sibility to form cyclic compounds. Only single cycle
variant is possible with three
C
atoms, but in more
complex molecules the number of cycles can increase
significantly. Small cycles of three and four atoms are
chemically unstable, which justifies using additional
constraint that eliminate small cycles. Constraints on
the number of
H
atoms attached to
C
may automati-
cally eliminate cycles through the valencyconstraints.
Including the MF to generate complete solutions
increase the ambiguity for amino acids further (Table
2) because it introduced
N
and
O
atoms not detected
in the NMR experiments. As the result, only valency
constraints can be formulated for these atoms, allow-
ing them to take any position in the molecule. How-
ever, for molecules that only contain
C
and
H
atoms
the MF constraints maybe highly beneficial because
they would eliminate structures with the incorrect to-
tal number of
H
atoms.
Our analysis highlights intrinsic ambiguity of
the NMR data and critical contributions from addi-
tional constraints for unambiguous determination of
the molecular structure. In the majority of CASE
systems, including the CS system of (Omrani and
Naanaa, 2016) the MF is required, and these con-
straints are rigidly embedded into the system; very
often the software would not run if these parameters
are not defined. This limits the usability of the sys-
tems, as most of the time only partial information on
these parameters is available at best. It may also lead
to incomplete or incorrect results, as possible solu-
tion are not explored systematically. In contrast, our
system explicitly defines all constraints used in find-
ing the solution and allows to explore the constraints
systematically. The system can be used with any set
of constraints, often incomplete, which allows its ap-
plication at all stages of the NMR analysis. The in-
spection of the results, as outlined above, can suggest
further experiments to eliminate ambiguities, until a
unique solution is found.
From a more theoretical perspective, we propose
to consider the obtained results in the complete set-
ting (including the formula) as evidence of the lim-
its of the NMR method itself. Indeed, in a consid-
ered variant of NMR analysis including HSQC and
HMBC spectra, even in the presence of all theoret-
Table 2: Complete solutions calculated for amino acids us-
ing NMR data and the MF as the inputs.
Amino acid MF Structures
# Sols Time (s) Nonisomorphic Time (s)
Alanine C
3
H
7
NO
2
662 0.10 148 0.00
Cysteine C
3
H
7
NO
2
S 8081 1.51 779 0.01
Glycine C
2
H
5
NO
2
78 0.00 34 0.00
Leucine C
6
H
13
NO
2
4091 2.41 136 0.00
Proline C
5
H
9
NO
2
15066 3.69 764 0.03
Serine C
3
H
7
NO
3
8081 1.51 783 0.01
Threonine C
4
H
9
NO
3
24153 5.81 732 0.04
Valine C
5
H
11
NO
2
2654 0.81 64 0.00
ically possible ideal NMR constraints (derived from
the known structure) the target structure can not be
identified uniquely. The numbers produced measure
an inherent ambiguity of the NMR method. We sus-
pect that it remains the case even in the presence of
further realistic constraints. The proposed CS-based
approach addresses such questions systematically and
this is a subject of our ongoing and future work.
6 RELATED WORK
The current study contributes to the existing knowl-
edge by addressing several structure elucidation prob-
lems. Unlike similar existing constrained structures
generating systems, the paper suggests an analytical
approach that considers being as realistic and practi-
cal as possible. As a result, the program generates
all possible structures based on real-world constraints,
considering the minimum available information.
There are a set of limitations that may affect the
molecular structure elucidation results. One poten-
tial drawback that might be encountered is the un-
certainty of NMR data, caused by the different envi-
ronments, especially during the acquisition of NMR
data, for solvent or temperature, which can create
significant noise of the spectra data. Further im-
provements should be taken into account to tackle
these problems. In the literature, several approaches
have been proposed to develop CASE systems over
the past century. The Dendral project (Smith et al.,
1981), is a pioneer in structure elucidation systems
based on NMR spectra. The main aim of the Den-
dral group was to develop computer systems to assist
the chemists in identifying unknown chemical struc-
tures (Gray, 1988). The project’s main achievement
was to introduce the CASE systems’ idea to eluci-
date the chemical structure. There has been a sig-
nificant increase in publications that document other
CASE systems’ approaches due to the development
of NMR techniques. The examples of these CASE
systems are SENECA platform-independent pack-
age (Steinbeck, 2001), MONOREG (Ferreira et al.,
Molecular Fragments from Incomplete, Real-life NMR Data: Framework for Spectra Analysis with Constraint Solvers
839
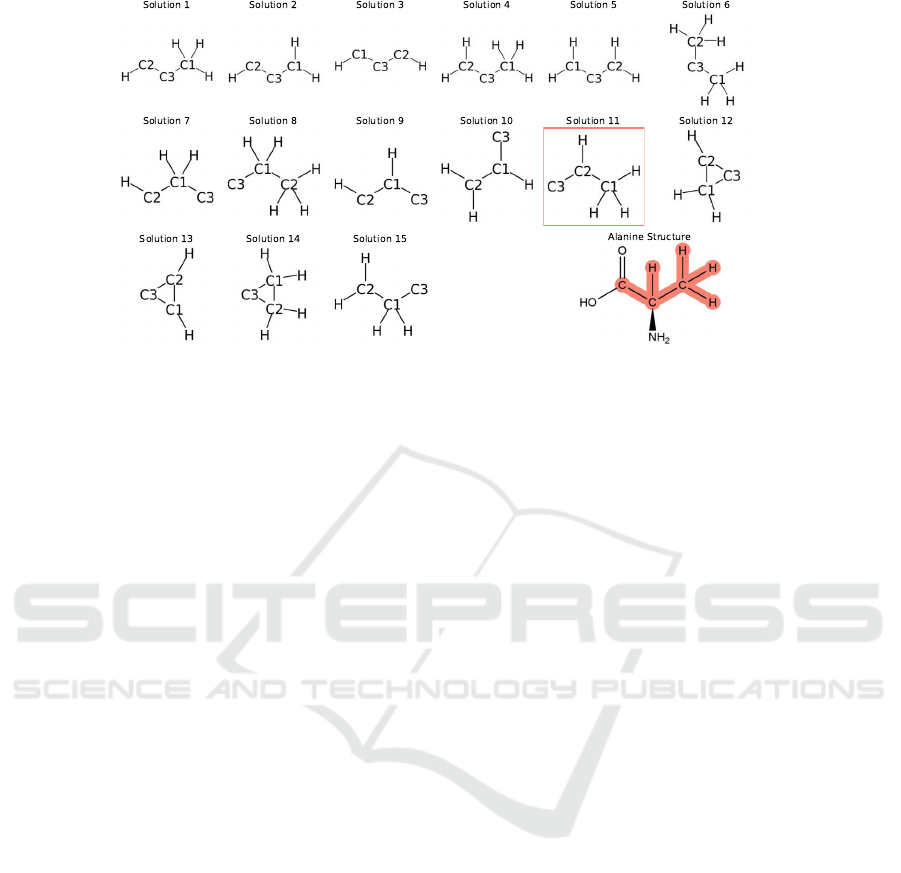
Figure 3: Solution multigraphs built from chemical and NMR constraints derived from alanine data. 15 multigraphs satisfied
the constraints and were connected and nonisomorphic. The box labelled (Solution 11) is the correct structure. The structure
at the bottom right is the complete structure of alanine and the highlighted parts in red corresponds to Solution 11.
2001), Bruker CMC-se program (Kessler and Gode-
johann, 2018), Logic for Structure Determination
(LSD) (Plainchont et al., 2013), Advanced Chem-
istry Development (ACD)/ Structure Elucidator Suite
(Elyashberg et al., 2002), Mestrelab MNova struc-
ture (Burns et al., 2019) and MOLGEN 5.0 (Gugisch
et al., 2015). The majority of these approaches rely
on complex sets of rules defined by experts, and spe-
cialised algorithms, which limits their effectiveness
and adaptation for complex systems, such as molecu-
lar mixtures. The constraint-based systems, we argue
for in this paper, separate rules (constraints) formula-
tion and constraint solving, which make them much
more flexible and adaptable. Such systems allow util-
ising the performance of generic constraint solvers.
According to (Omrani and Naanaa, 2016), the frame-
work of CS provides an effective solution to structure
elucidation problems. A program was developed to
generate molecular structures based on satisfying sev-
eral predefined constraints. The first constraint is the
MF. In addition, the number of bonds for each atom
and the type of bonds are given as inputs to their sys-
tem. The system can also impose substructures to ap-
pear in the results and forbid specific fragments to ex-
ist in the generated structures. The authors have pub-
lished a recent study describing a new open-source
system CP-MolGen (Omrani and Naanaa, 2019).
However, from a practical perspective, the amount
of minimum required information to use the frame-
work proposed by the authors is challenging to ob-
tain and even unfeasible in some cases. The MFs are
not always readily available. Determining the num-
ber of bonds may require extensive analytical data
from different instruments. A detailed map of spe-
cific distance between atoms is a complicated process
that usually is incomplete. For example, HMBC ex-
periments are used to map out the bond distances but
HMBC experiments cannot distinguish between two
or three bonds (Janovick et al., 2020). This infor-
mation needs to be inferred from other data available
similar to a CS approach. In practice, after obtaining
such detailed data elucidating the molecular structure
is a trivial process and using a constraint solver is gen-
erally not necessary.
Structure elucidation can be formulated as a more
generalised CS problem, and approximate solutions
can be proposed with a more realistic amount and
type of input information. All constraints can be for-
mulated exactly and explicitly, which allows avoiding
biased solutions. Such a CS approach to obtain a list
of approximations down to the least ambiguous set
would be more useful in practical applications. Such a
list of solutions can even indicate which experiments
to perform next to get a unique solution. In a typi-
cal NMR analysis, samples usually consist of chemi-
cal mixtures or contain contaminants that have signals
indistinguishable from the compounds of interest.
Thus, the amount of experimental NMR informa-
tion is very limited, which leads to incomplete con-
nectivity and a lack of separation of the observed sig-
nals into groups corresponding to different molecules.
Sometimes partial information on the MFs can be ob-
tained from the MS analysis, but cannot be related
to the NMR signals. These challenges are impossi-
ble to address with the reported CS implementation
of (Omrani and Naanaa, 2016). The CASE system
we have presented addresses this particular need and
allows obtaining a full list of possible chemical struc-
tures irrespective of the completeness of the NMR in-
formation. This creates a powerfulcomputational tool
ICAART 2022 - 14th International Conference on Agents and Artificial Intelligence
840

for NMR data analysis and guidance for the selection
of additional experiments to establish an objectively
unique solution.
7 CONCLUSIONS
This paper presents a method of interpreting NMR
spectra data via the CS framework to generate molec-
ular multigraphs. Real-world chemical structures in-
stances demonstrate that obtaining constraints based
on NMR data is successfully predicts the correct
molecular multigraphs. While alternativeCS methods
exist, which can determine the correct structures for
the molecules based on several defined constraints,
the interpretation of spectra data in our approach has
the advantage of generating the structures based on
the minimum available information as the case of the
real practical NMR instances. Although we solve
the structures predicting problems, we did not con-
sider more complex natural compounds. For instance,
chemical structures containing ring compounds and
the existence of symmetry for some atoms of the
structures. The main challenge would be that it can
not easily differentiate between the atoms as two or
more atoms will be had the same values of chemical
shifts. Further measures are suggested to improve the
performance and to predict robust and confident struc-
tures. The actions should define the most appropriate
methods to handle any uncertainty of NMR data as
this is more likely to be encountered in real instances
of spectra data.
REFERENCES
Burns, D. C., Mazzola, E. P., and Reynolds, W. F. (2019).
The role of computer-assisted structure elucidation
(case) programs in the structure elucidation of com-
plex natural products. Natural product reports,
36(6):919–933.
Elyashberg, M. and Williams, A. J. (2015). Computer-
based structure elucidation from spectral data.
Springer.
Elyashberg, M. E., Blinov, K. A., Williams, A. J., Mar-
tirosian, E. R., and Molodtsov, S. G. (2002). Appli-
cation of a new expert system for the structure eluci-
dation of natural products from their 1d and 2d NMR
data. Journal of natural products, 65(5):693–703.
Ferreira, M. J., Rodrigues, G. V., and Emerenciano, V. P.
(2001). Monoreg an expert system for structural elu-
cidation of monoterpenes. Canadian journal of chem-
istry, 79(12):1915–1925.
Gray, N. (1988). Dendral and meta-dendral—the myth and
the reality. Chemometrics and Intelligent Laboratory
Systems, 5(1):11–32.
Gugisch, R., Kerber, A., Kohnert, A., Laue, R., Meringer,
M., R¨ucker, C., and Wassermann, A. (2015). Molgen
5.0, a molecular structure generator. In Advances in
mathematical chemistry and applications, pages 113–
138. Elsevier.
Hebrard, E. (2008). Mistral, a constraint satisfaction li-
brary. Proceedings of the Third International CSP
Solver Competition, 3(3):31–39.
Hebrard, E., O’Mahony, E., and O’Sullivan, B. (2010).
Constraint programming and combinatorial optimisa-
tion in numberjack. In Lodi, A., Milano, M., and Toth,
P., editors, AI and OR Techniques in Optimization
Italy, June 14-18, 2010. Proceedings, volume 6140 of
Lecture Notes in Computer Science, pages 181–185.
Springer.
Janovick, J., Spyros, A., Dais, P., and Hatzakis, E. (2020).
4 - nuclear magnetic resonance. In Pico, Y., editor,
Chemical Analysis of Food (Second Edition), pages
135–175. Academic Press, second edition edition.
Kessler, P. and Godejohann, M. (2018). Identification
of tentative marker in corvina and primitivo wines
with cmc-se. Magnetic Resonance in Chemistry,
56(6):480–492.
Koichi, S., Arisaka, M., Koshino, H., Aoki, A., Iwata, S.,
Uno, T., and Satoh, H. (2014). Chemical structure
elucidation from 13c NMR chemical shifts: Efficient
data processing using bipartite matching and maximal
clique algorithms. Journal of chemical information
and modeling, 54(4):1027–1035.
Landrum, G. (2016). Rdkit: Open-source cheminformatics
software. GitHub and SourceForge, 10:3592822.
McKay, B. D. and Piperno, A. (2014). Practical graph
isomorphism, ii. Journal of symbolic computation,
60:94–112.
Omrani, M. A. and Naanaa, W. (2016). A constrained
molecular graph generation with imposed and forbid-
den fragments. In Proceedings of the 9th Hellenic
Conference on Artificial Intelligence, pages 1–5.
Omrani, M. A. and Naanaa, W. (2019). Constraints for gen-
erating graphs with imposed and forbidden patterns:
an application to molecular graphs. Constraints, pages
1–22.
Plainchont, B., de Paulo Emerenciano, V., and Nuzillard,
J.-M. (2013). Recent advances in the structure eluci-
dation of small organic molecules by the lsd software.
Magnetic Resonance in Chemistry, 51(8):447–453.
Smith, D. H., Gray, N. A., Nourse, J. G., and Crandell,
C. W. (1981). The dendral project: Recent advances
in computer-assisted structure elucidation. Analytica
Chimica Acta, 133(4):471–497.
Steinbeck, C. (2001). Seneca: A platform-independent,
distributed, and parallel system for computer-assisted
structure elucidation in organic chemistry. Jour-
nal of chemical information and computer sciences,
41(6):1500–1507.
Ulrich, E. L., Akutsu, H., Doreleijers, J. F., Harano, Y.,
Ioannidis, Y. E., Lin, J., Livny, M., Mading, S., Maz-
iuk, D., Miller, Z., et al. (2007). Biomagresbank. Nu-
cleic acids research, 36(suppl
1):D402–D408.
Molecular Fragments from Incomplete, Real-life NMR Data: Framework for Spectra Analysis with Constraint Solvers
841
