
One Size Does Not Fit All: An Ensemble Approach Towards Information
Extraction from Adverse Drug Event Narratives
Susmitha Wunnava
1,∗
, Xiao Qin
1,∗
, Tabassum Kakar
1,∗
, Xiangnan Kong
1
, Elke A. Rundensteiner
1
,
Sanjay K. Sahoo
2
and Suranjan De
2
1
Computer Science Department, Worcester Polytechnic Institute, Worcester, MA, U.S.A.
2
Center for Drug Evaluation and Research, U.S. Food and Drug Administration, Silver Spring, MD, U.S.A.
Keywords:
Pharmacovigilance, Adverse Drug Reaction, Class Imbalance, Ensemble Learning.
Abstract:
Recognizing named entities in Adverse Drug Reactions narratives is a fundamental step towards extracting
valuable patient information from unstructured text into a structured thus actionable format. This then un-
locks advanced data analytics towards intelligent pharmacovigilance. Yet existing biomedical named entity
recognition (NER) tools are limited in their ability to identify certain entity types from these domain-specific
narratives and result in significant performance differences in terms of accuracy. To address these challenges,
we propose an ensemble approach that integrates a rich variety of named entity recognizers to procure the final
result. First, one critical problem faced by NER in the biomedical context is that the data is highly skewed.
That is, only 1% of words belong to a certain medical entity type, such as, the reason for medication usage
compared to all other non-reason words. We propose a balanced, under-sampled bagging strategy that is de-
pendent on the imbalance level to overcome the class imbalance problem. Second, we present an ensemble
of heterogeneous recognizers approach that leverages a novel ensemble combiner. Our experimental results
show that for biomedical text datasets: (i) a balanced learning environment along with an Ensemble of Hetero-
geneous Classifiers constantly improves the performance over individual base learners and, (ii) stacking-based
ensemble combiner methods outperform simple Majority Voting by 0.30 F-measure.
1 INTRODUCTION
1.1 Motivation and Background
Adverse Drug Reactions (ADRs) correspond to an un-
wanted and often extremely dangerous effect caused
by the administration of drugs. ADRs unrevealed du-
ring the clinical trials are one of the leading causes of
death worldwide (Lazarou et al., 1998). To oversee
the safety and effectiveness of the drugs in the post
marketing phase, surveillance systems such as FDA
Adverse Event Reporting System (FAERS) monitor
the ADR incidences submitted by consumers, healt-
hcare professionals and drug manufacturers. These
reports are reviewed by FDA staff to identify potential
∗
Susmitha Wunnava is thankful to the Seeds of STEM
and Institute of Education Sciences, U.S. Department of
Education for supporting her PhD studies via the grant
R305A150571. Xiao and Tabassum are grateful to Oak
Ridge Associated Universities (ORAU) for granting them
an ORISE Fellowship to conduct research with the U.S.
Food and Drug Administration.
drug safety concerns and, when necessary, to recom-
mend appropriate actions to improve product safety.
In 2015, over 1.7 million of incidents are repor-
ted to FAERS and the number is growing making the
drug review process more challenging (FDA, 2016).
To effectively identify drug safety signals in a timely
manner from the exploding amount of reports with li-
mited human resources, the reviewing processes are
enhanced by advanced data mining and visualization
technologies (Wilson et al., 2004; Feng et al., 2013;
Sakaeda et al., 2013). However, most of these techno-
logies rely on information organized in structured for-
mat where the unstructured text has to be first proces-
sed and converted into structured information.
Although the original report has structured fields,
the unstructured narratives in the MedWatch form
used for reporting an adverse event (Illustrated in Fig.
2) often contain information that is left blank in the
structured fields. More importantly, these narratives
are rich in detailed information regarding the adverse
event as shown in Fig.1. Automatically extracting in-
formation from the unstructured ADR report narrati-
176
Wunnava, S., Qin, X., Kakar, T., Kong, X., Rundensteiner, E., Sahoo, S. and De, S.
One Size Does Not Fit All: An Ensemble Approach Towards Information Extraction from Adverse Drug Event Narratives.
DOI: 10.5220/0006600201760188
In Proceedings of the 11th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2018) - Volume 5: HEALTHINF, pages 176-188
ISBN: 978-989-758-281-3
Copyright © 2018 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

ves into structured format is critical for advanced ana-
lytics and vital for timely detection, assessment and
prevention of future incidents of ADRs. In this study,
we focus on the Named Entity Recognition (NER) –
a fundamental task in this process, to classify the in-
formation categories in the narratives.
A major hurdle with biomedical narratives especi-
ally with processing medical reports is that the text is
unstructured, comprised of different formats and sty-
les depending upon the report source. First, a named
entity phrase could be expressed as a combination of
entity-specific medical terms as well as non-medical
descriptive text. For instance, in the named entity
phrase “coronary artery disease related event prophy-
laxis”, the words “related” and “event” are descriptive
text while the rest are medical terms. Named entity
phrases such as these can cause ambiguity even du-
ring the manual annotation process. Second, the nar-
ratives are predominately composed of large chunks
of texts with sparse relevant phrases specific to the
named entities.
Given above observations, it is a common proto-
col to engage multiple expert annotators specializing
in different types of biomedical text and specific types
of named entity to recognize and tag phrases and, then
as a final step combine their expert opinions to come
to an inter-expert agreement for determining the final
output. As shown in our experiments, this problem
persists when it comes to automatically recognizing
entities through computational approaches. A named
entity recognizer for biomedical text is usually desig-
ned for specific text type or entity type where a ge-
neric approach will almost certainly fail the domain
specific task. Recently, many biomedical NER sys-
tems (Xu et al., 2010; Aronson, 2001; Uzuner et al.,
2010a; Savova et al., 2010) and frameworks (Ferrucci
and Lally, 2004) have been proposed customized for
specific domain and entity type. To the best of our
knowledge, there is no study today on how to auto-
matically adapt and integrate the strength of a rele-
vant and yet diverse set of named entity recognizers
to tackle a new domain specific NER task.
1.2 Related Work
Existing approaches to biomedical NER can be cate-
gorized into rule-based, machine learning based and
hybrid methods.
The rule-based methods leverage user-defined pat-
tern matching rules supported with semantic know-
ledge resources. MedLEE (Friedman et al., 1994) and
MedEx (Xu et al., 2010) are rule-based systems that
use a medical knowledge base and a linguistic ap-
proach to extract relevant medical information from
clinical text. While rule-based systems perform well
on identifying known patterns, they are limited in
their ability to generalize. They thus fail to identify
unknown words and patterns.
Machine learning based methods learn from fea-
tures extracted from words and thus have a better ge-
neralization ability compared to rule-based methods.
However, they require large annotated corpora for
training. (Uzuner et al., 2009) demonstrated that ma-
chine learning approaches can outperform rule-based
systems for assertion classification in clinical text.
(Ramesh et al., 2014) developed a biomedical named
entity tagger using Support Vector Machines (SVM)
to extract medication and ADR information from FA-
ERS narratives. (Ghiasvand, 2014) used Conditio-
nal Random Fields (CRF) to label diseases and dis-
orders in clinical sentences. (Halgrim et al., 2011)
used a Maximum Entropy model to extract relevant
medical information. (Jagannatha and Yu, 2016) used
Recurrent Neural Networks to extract medical events
from Electronic Health Records (EHR) and showed
that they significantly outperformed the CRF models.
Hybrid approaches that utilize both rule-based and
machine learning methods have also began to be ex-
plored. (Doan and Xu, 2010) developed an SVM
based method that utilizes the semantic tags of the
words obtained from MedEx as features to recog-
nize medication-related entities from discharge sum-
maries.
1.3 Challenges of Entity Recognition
using Machine Learning
The focus of our research is on supervised machine
learning methods for biomedical NER and classifica-
tion. In particular, we focus on a two-class, binary
classification task to recognize and classify named en-
tities. Despite its value and significance, biomedical
NER and classification is a more challenging task due
to the specific characteristics of the task. Two of the
most critical challenges are:
1. Lack of Positive Class Instances & Class Imba-
lance: One problem in classifying named entities
in biomedical text especially clinical text is that the
data in the training dataset is predominately compo-
sed of non-medical text with only a small percentage
of entity-specific medical text leading to highly ske-
wed and imbalanced class distributions. Usually, the
positive class, i.e., the class of interest that represents
the named entity, has very few instances and is in a
stark minority compared to the negative class (e.g.,
reason vs non-reason instances in the narratives, see
Fig. 1).
One Size Does Not Fit All: An Ensemble Approach Towards Information Extraction from Adverse Drug Event Narratives
177
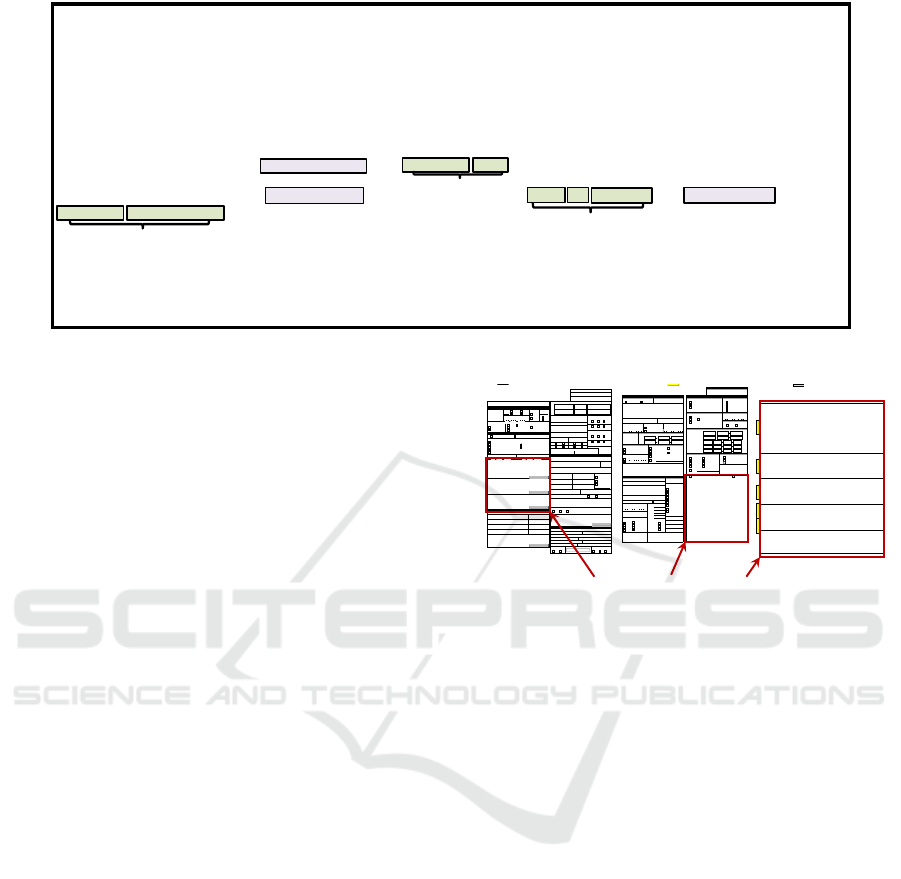
FDA Adverse Event Reporting System (FAERS)
Individual Case Safety Reports (ICSRs)
Event Narrative
This spontaneous report from a patient concerns a XX-year-old Caucasian female from the United States: Local
ID: X-XXXXXXXXXX.
The patient's weight was XXX pounds and height was XX.X inches. Concurrent conditions included abdominal
bloating, abdominal gas, diabetic paresis and type 2 diabetes. The patient had previously experienced
allergy when taking mycins ( a
ntibacterials
for systemic use )and sulfa.
The patient was treated with
canagliflozin
!!
300 mg once a day for type 2 diabetes and domperidone
for
diabetic gastroparesis .
In XXX-XXXX, the patient contacted her physician about the events and was prescribed an increased dosage of
domperidone. The dose of domperidone was increased and the dose of canagliflozin was not changed. The
patient reported the increased dose of domperidone had not relieved her worsening symptoms. The patient had
not recovered from increased belching, worsening of abdominal pain, very gaseous, worsening of bloating, and
not feeling well today. This report was identified by the call center as a product quality complaint.
gastroparesis!diabetic!
systemic! use!
type! 2!
diabetes!
antibacterials!
domperidone!
canagliflozin!
[REASON])
[MEDICATION]!
[REASON])
[REASON])
[MEDICATION]!
[MEDICATION]!
Figure 1: A sample FAERS report highlighting detailed information on the ADR incident within the narrative.
Research (Longadge and Dongre, 2013; Japko-
wicz and Stephen, 2002) has found that, learning on
imbalanced training datasets can cause a significant
deterioration in the performance of the supervised
machine learning methods, particularly when clas-
sifying instances belonging to the under-represented
class.
2. Lack of a Single Best Performing Classification
Method: It is challenging to choose the appropri-
ate learning algorithm to train and classify the new
instances. Conventional approaches to biomedical
NER tend to use a single machine learning method
such as Support Vector Machines (SVM), Conditio-
nal Random Fields (CRF), Maximum Entropy (ME)
(Bishop, 2006) classify named entities in the text.
Each of these methods have some advantages over
the others and differs significantly in their performan-
ces in classifying the named entities. (Uzuner et al.,
2010a) shows that the teams that used different su-
pervised machine learning methods on the same da-
taset obtained significantly different results from one
another. Additionally, the performances of a single
system across the various named entities is shown to
differ. (Uzuner et al., 2010a) concluded that although
the state-of-the-art NLP systems perform well in ex-
tracting some of the named entities (such as medica-
tion, dosages), while other entities (duration, reason
for administration) have shown to be very challen-
ging.
1.4 The Scope of this Work
The general problems of class imbalance and ensem-
ble learning systems for classification have been stu-
died in the literature (Galar et al., 2012). However, in
the context of biomedical NER, a collective approach
to deal with both the class imbalance problem and the
U.S. Department of Health and Human Services
Food and Drug Administration
B. ADVERSE EVENT OR PRODUCT PROBLEM
D. SUSPECT MEDICAL DEVICE
E. INITIAL REPORTER
Form Approved: OMB No. 0910-0291, Expires: 9/30/2018
See PRA statement on reverse.
FORM FDA 3500A (10/15)
Submission of a report does not constitute an admission that medical
personnel, user facility, importer, distributor, manufacturer or product
caused or contributed to the event.
MEDWATCH
For use by user-facilities,
importers, distributors and manufacturers
for MANDATORY reporting
UF/Importer Report #
FDA Use Only
Mfr Report #
PLEASE TYPE OR USE BLACK INK
1.
5. Describe Event or Problem
2. Common Device Name
11. Concomitant Medical Products and Therapy Dates
(Exclude treatment of event)
3. Manufacturer Name, City and State
6. If Implanted, Give Date
(dd-mmm-yyyy)
2. Health
Professional?
8. Is this a single-use device that was
reprocessed and reused on a patient?
9. If Yes to Item 8, Enter Name and Address of Reprocessor
10. Device Available for Evaluation? (Do not send to FDA)
4. Model #
Catalog #
Serial #
6. Relevant Tests/Laboratory Data, Including Dates
1. Brand Name
7. Other Relevant History, Including Preexisting Medical Conditions (e.g.,
allergies, pregnancy, smoking and alcohol use, liver/kidney problems, etc.)
2b. Procode
Lot #
Expiration Date
(dd-mmm-yyyy)
Unique Identifier (UDI) #
5. Operator of Device
7. If Explanted, Give Date
(dd-mmm-yyyy)
3. Occupation (Select from list)
4. Initial Reporter Also Sent
Report to FDA
Adverse Event
and/or
Product Problem (e.g., defects/malfunctions)
Yes
Yes
Yes Yes
No
No Returned to Manufacturer on:
No No Unk
Health
Professional
Lay User/Patient
Other
(Continue on page 3)
(Continue on page 3)
(Continue on page 3)
(Continue on page 3)
A. PATIENT INFORMATION
1. Patient Identifier
In Confidence
2. Age
or Date of Birth (e.g., 08 Feb 1925)
3. Sex 4. Weight
Female
Year(s) Month(s)
Days(s)Week(s)
Male
lb
kg
Note: For date prompts of “dd-mmm-yyyy” please use 2-digit day, 3-letter month
abbreviation, and 4-digit year; for example, 01-Jul-2015.
2. Outcome Attributed to Adverse Event (Check all that apply)
3. Date of Event (dd-mmm-yyyy) 4. Date of this Report (dd-mmm-yyyy)
Death
Life-threatening Disability or Permanent Damage
Congenital Anomaly/Birth DefectsHospitalization – initial or prolonged
Other Serious (Important Medical Events)
Required Intervention to Prevent Permanent Impairment/Damage (Devices)
C. SUSPECT PRODUCT(S)
1. Name, Manufacturer/Compounder, Strength
#1 – Name and Strength
#2 – Name and Strength
2. Concomitant Medical Products and Therapy Dates (Exclude treatment of event)
#1 – NDC # or Unique ID
#2 – NDC # or Unique ID
#1 – Manufacturer/Compounder
#2 – Manufacturer/Compounder
#1 – Lot #
#2 – Lot #
4. Therapy Dates (If unknown, give duration) from/
to (or best estimate)) (dd-mmm-yyyy)
5. Diagnosis for Use (Indication)
8. Expiration Date (dd-mmm-yyyy)
9. Event Abated After Use
Stopped or Dose Reduced?
10. Event Reappeared After
Reintroduction?
Yes
Yes
Yes
Yes
No
No
No
No
Doesn’t
apply
Doesn’t
apply
Doesn’t
apply
Doesn’t
apply
#1
#1
#1
#2
#2
#1 #2
#1
#2
#2
- -
- -
- -- -
- -- -
- -
1. Name and Address
Last Name:
Address:
City:
Country:
Phone #:
First Name:
State/Province/Region:
Email:
ZIP/Postal Code:
5.a. Ethnicity (Check
single best answer)
Hispanic/Latino
Not Hispanic/Latino
5.b. Race (Check all that apply)
Asian American Indian or Alaskan Native
Black or African American White
Native Hawaiian or Other Pacific Islander
Include date (dd-mmm-yyyy):
- -
6. Is the Product
Compounded?
7. Is the Product Over-
the-Counter?
Yes Yes
Yes Yes
No No
No No
#1 #1
#2 #2
3.
Dose Frequency Route Used
#1
#2
Page 1 of 3
-- --
(Continue on page 3)
Reset Form
(Continue on page 3)
(Continue on page 3)
(Continue on page 3)
(Continue on page 3)
(Continue on page 3)
G. ALL MANUFACTURERS
Health Professional
2. Phone Number
1. Contact Office (and Manufacturing Site for Devices)
Name
3. Report Source
(Check all that apply)
Literature
Foreign
Study
Consumer
Email Address
Compounding Outsourcing Facility 503B?
Address
Yes
Company
Representative
User Facility
Distributor
Other:
ANDA #
10.
Additional Manufacturer Narrative
11.
Corrected Data
and / or
F. FOR USE BY USER FACILITY/IMPORTER (Devices Only)
FDA USE ONLY
H. DEVICE MANUFACTURERS ONLY
3. User Facility or Importer Name/Address
1. Check One 2. UF/Importer Report Number
User Facility
4. Contact Person
6. Date User Facility or
Importer Became Aware
of Event (dd-mmm-yyyy)
(Specify)
7. Type of Report
9. Approximate
Age of Device
Device
Code
10. Event Problem Codes (Refer to coding manual)
Patient
Code
11. Report Sent to FDA? (If Yes,
enter date (dd-mmm-yyyy))
8. Date of This Report
(dd-mmm-yyyy)
12. Location Where Event Occurred
14. Manufacturer Name/Address
5. Phone Number
13. Report Sent to Manufacturer? (If
Yes, enter date (dd-mmm-yyyy))
Outpatient
Diagnostic Facility
Outpatient Treatment
Facility
Conclusions
Nursing Home
Follow-up #
Initial
Other:
NDA #
No
Yes
Evaluation Summary Attached
Home
8. Adverse Event Term(s)
30-day
6. If IND, Give Protocol #
7. Type of Report
(Check all that apply)
4. Date Received by
Manufacturer (dd-mmm-yyyy)
5.
Combination
Product
No
9. Manufacturer Report Number
Yes
5-day
Modification/
Adjustment
7-day
Yes
Yes
10-day
Periodic
PMA/
510(k) #
1. Type of Reportable Event 2. If Follow-up, What Type?
Ambulatory
Surgical Facility
3. Device Evaluated by Manufacturer?
4. Device Manufacture Date
(dd-mmm-yyyy)
5. Labeled for Single Use?
No (Attach page to explain why not) or
provide code:
Importer
No
6. Event Problem and Evaluation Codes (Refer to coding manual)
7. If Remedial Action Initiated, Check Type
Method
9. If action reported to FDA under
21 USC 360i(f), list correction/
removal reporting number:
8. Usage of Device
Yes
Death
Hospital
Notification
Inspection
Patient Monitoring
Results
Yes
Not Returned to Manufacturer
Serious Injury
Malfunction
Relabeling
Correction
Additional Information
Response to FDA Request
Device Evaluation
Initial Use of Device
Reuse
Unknown
Other:
Recall
Repair
Replace
15-day
Pre-1938
BLA #
IND #
FORM FDA 3500A (10/15) (continued)
MEDWATCH
Initial
Follow-up # ____
Device
Code
Patient
Code
Yes
OTC
Department of Health and Human Services
Food and Drug Administration
Office of Chief Information Officer
Paperwork Reduction Act (PRA) Staff
PRAStaff@fda.hhs.gov
This section applies only to requirements of the Paperwork Reduction Act of 1995.
The public reporting burden for this collection of information has been estimated to average 73
minutes per response, including the time for reviewing instructions, searching existing data
sources, gathering and maintaining the data needed, and completing and reviewing the collection
of information. Send comments regarding this burden estimate or any other aspect of this
collection of information, including suggestions for reducing this burden to:
Please DO NOT RETURN this form to the above PRA Staff email address.
OMB Statement: "An agency may not
conduct or sponsor, and a person is not
required to respond to, a collection of
information unless it displays a currently
valid OMB control number."
- - - -
- -
- -
- -
- -
Page 2 of 3
Reset Form
(CONTINUATION PAGE)
For use by user-facilities,
importers, distributors, and manufacturers
for MANDATORY reporting
Concomitant Medical Products and Therapy Dates (Exclude treatment of event) (For continuation of C.2 and/or D.11; please distinguish)
Other Remarks
B.5. Describe Event or Problem (continued)
B.6. Relevant Tests/Laboratory Data, Including Dates (continued)
B.7. Other Relevant History, Including Preexisting Medical Conditions (e.g., allergies, pregnancy, smoking and alcohol use, hepatic/renal dysfunction, etc.) (continued)
FORM FDA 3500A (10/15) (continued)
MEDWATCH
Page 3 of 3
Back to Item B.5
Back to Item B.6
Back to Item B.7
Back to Item C.2
Back to Item D.11
Reset Form
Report Narrative (Free Text)
Figure 2: FAERS report – Medwatch 3500A.
limitations of any one individual classification met-
hod has not been studied extensively. In this paper,
we thus design a novel methodology called Tiered En-
semble Learning System with Diversity (TELS-D) to
address the above challenges in NER. TELS-D invol-
ves four core steps: 1) To address the class imbalance
inherent in medical data used for machine learning
training, we create a balanced training environment
by applying undersampling techniques. 2) We gene-
rate an ensemble of diverse classifiers by training a set
of heterogeneous learning algorithms in this balanced
training environment. 3) We combine the intermedi-
ate results generated by each of the classifiers in the
ensemble to create a meta-training feature set. 4) We
train a “learner-over-learners” meta-algorithm over
the meta-level features to correctly learn and classify
the named entities in the narratives.
To evaluate our model, we perform comprehen-
sive experiments on biomedical reports datasets. Our
experiments demonstrate that our proposed methodo-
logy TELS-D outperforms the individual learners in
the ensemble. TELS-D achieves a higher accuracy
of 0.52 F-measure compared to any of the individual
classifiers with F-measure ranging from 0.22-0.33, in
recognizing the relevant information categories from
the narratives.
HEALTHINF 2018 - 11th International Conference on Health Informatics
178

2 METHODOLOGY
2.1 The Data Set
The FDA FAERS Adverse Event Report Narrati-
ves. The FDA Adverse Event Reporting System (FA-
ERS) is a database that contains information on ad-
verse events and medication errors in the form of re-
ports submitted to the FDA from various sources such
as patients, medical professionals and drug manufac-
turers. A report contains both a structured section of
content followed by some free-form text. Fig. 2 de-
picts an example of MedWatch report form supported
by FAERS. As many studies indicate (Harpaz et al.,
2014), the narrative can be either supplementary ma-
terial to the structured fields or in many cases repor-
ters tend to provide a detailed narrative in the unstruc-
tured format without taking the effort to fill in all the
structured fields. Therefore, there is a need for iden-
tifying information related to the adverse event case
from the free text in order to collect all relevant kno-
wledge about the case in structured and thus a easy
processable format.
In this study, we aim to identify one important
piece of knowledge, namely the reason thought to be
the cause of the administration of the medication as
per the FAERS report narrative. While we work with
925 FAERS reports, they are unlabeled and not re-
dacted and therefore not available to the general pu-
blic due to patient’s privacy concerns. In addition, we
also work with 16 redacted reports provided by the
FDA as briefly described in Table 1.
Table 1: Statistics for the datasets.
FAERS i2b2
#Reports 16 242
#Sentences 678 8,050
#All Words 6,116 67,074
#Reason Words NA 1,881
Data Set of Annotated Patient Discharge Sum-
maries by Partners Healthcare. To assure reprodu-
cibility, we also work with the publicly available data
set from the 2009 Medication Extraction Challenge
from the Third i2b2 Workshop on Natural Language
Processing Challenges for Clinical Records (Uzuner
et al., 2010b; Uzuner et al., 2010a). The data set con-
sists of annotated patient discharge summaries pro-
vided by Partners Healthcare. As part of the chal-
lenge, 696 reports were released for training out of
which 17 reports were annotated by the i2b2 orga-
nizers. An additional 251 reports were released as
the testing data set and were annotated by the partici-
pating teams. Annotated entities include medication
name, dosage, mode, frequency, duration, and reason
for administration. We work with 242 annotated re-
ports (9 from the annotated training set and 233 from
the testing set) as described in Table 1.
In this work, we focus on identifying the reason
entity for the administration of drug from these dis-
charge summaries. First, the reason entity has routi-
nely been pointed out as one of the important fields
yet among the hardest to recognize and extract due
to its diversity and often not well scoped vocabu-
lary (Uzuner et al., 2010a; Halgrim et al., 2011). The
original dataset features a heavy class imbalance with
respect to the reason type. That is, tokens labeled as
belonging to the reason class represent about 1% of
all the tokens in these reports. Since the goal of this
study is to develop an information extraction strategy
that successfully identifies the reason for administra-
tion from the text, we focus on the narrative section
of each report.
2.2 Data Pre-Processing
Data pre-processing is vital for converting the raw
textual data into a processable format suitable for the
natural language processing. We use following steps
to pre-process each report in the corpus:
1. Sentence Segmentation: Each report is split into
sentences to decompose the structure.
2. Word Tokenization: Each sentence is split into to-
kens (words) as this is our unit of processing.
3. Punctuation Removal: All tokens that represent
punctuations are removed.
2.3 Feature Extraction
A rich set of features are needed for machine learning
to learn the meaning of tokens. For each word token
obtained from the preprocessing module we generate
the following feature sets:
1. Word Features: The token is converted into a bag-
of-words representation based on the vocabulary of
the entire corpus. To generate the vocabulary, words
in the corpus are converted to lowercase and stemmed
using the NLTK Porter Stemmer (Bird et al., 2009).
2. Syntactic Features: A constituency parse tree is
created using Charniak-Johnson parser (Charniak and
Johnson, 2005). Each token is tagged with its re-
spective parts-of-speech (POS) and lexical categories.
3. Semantic Features: Semantic categories of the
word are then obtained through lexicon lookup from
medication lexicons, side effect lexicons (such as SI-
DER) (Kuhn et al., 2015) as well as UMLS Metamap
(Aronson, 2001).
One Size Does Not Fit All: An Ensemble Approach Towards Information Extraction from Adverse Drug Event Narratives
179

Token&< F
1
,F
2
…>
D
1
D
2
D
3
D
4
D
5
D
6
non-
reason
reason
non-
reason
reason
non-
reason
reason
non-
reason
(a) DT
Logistic(Regression
(b) LR
SVM
(c) SVM
Figure 3: Illustration of machine learning models.
4. Context Features: Words adjacent to the token in
the narrative provide the context in which the token
is actually used. This feature is helpful to differenti-
ate when a token falls into one of two different secti-
ons of a report and thus labeled differently. A con-
text window size five words i.e,. two words before
and two words after the token are coded using bag-
of-words representation. A boolean value is a binary
flag that indicates whether this token occurs before or
after certain so called “trigger words”. We identify
trigger words that may indicate the presence of the
named entity reason.
5. Morphological: The suffix and prefix of up to 3
characters within this word. For example: 1) words
with prefix of “dys” indicate something is abnormal,
such as dyspnea, 2) words with a suffix of “ing” may
indicate a condition or symptom, such as bloating.
6. Orthographic: Boolean values are used to indicate
if this word contains capital letters, digits, special cha-
racters, etc.
2.4 Base Machine Learning Models
After each token has been characterized by descrip-
tive features by the above step, the tokens in the form
of feature vector along with their associated label in-
dicating its class type (reason or non-reason) are then
used to train the models.
Different machine learning models have their own
set of assumptions and way of modeling the data, re-
sulting in its pros and cons in the classification task.
In our study, we assume that different models are able
to capture different aspects of the data and having
them compliment each other in an assembly fashion
will achieve better accuracy than any of them working
individually. We build our base classifiers using mul-
tiple popular machine learning models, namely, Deci-
sion Tree (DT), Logistic Regression (LR) and Support
Vector Machine (SVM) (Alpaydin, 2014) (Illustrated
in Fig. 3).
2.5 Ensemble of Classifiers
Ensemble of classifiers is a group of diverse classi-
fiers whose classification recommendations are aggre-
gated to achieve more accurate classification (Alpay-
din, 2014; Polikar, 2009). The goal of an ensemble
system is to combine the results of many diverse clas-
sifiers into a single consensus result that outperforms
any one of the individual classifiers by reducing their
generalization error and thus their misclassification
rate. The generalization error of the ensemble system
tends to be lower than that of the individual classi-
fiers when there is sufficient diversity in the ensem-
ble where the base learners have different prediction
accuracy on different instances. This makes the as-
sumption that the base learners are better performing
than random guessing. They have an accuracy greater
than 50% (Tan et al., 2006).
2.5.1 Ensemble Generation: Model Diversity
1. Heterogeneous Learning Methods:
One approach
to generating a diverse set of classifiers is to train
different learning methods on the same training set.
If the performance of each of these methods varies
significantly, then the results obtained are diverse in
nature. Then to overcome the limitations of each le-
arning algorithm while taking advantage of their re-
spective strengths is to combine the classifiers into
an ensemble of classifiers. In this study, we thus
follow this methodology and create an ensemble of
models obtained with the SVM, LR and DT learning
methods. Our experiments (see Sec.3.8) confirm that
an ensemble of these base classifiers outperforms any
one of them.
2. Heterogeneous Training Datasets: Another com-
mon approach to generating a diverse set of classifiers
is to create different subsets of the original training
dataset and then to train a single learning method on
each of the subsets from the training data set. Bag-
ging (Breiman, 1996) and Boosting (Schapire, 1990)
are examples of algorithms that tackle the generation
of collection of classifiers by sub-setting the original
dataset. However, given that our data set suffers from
a heavy class imbalance problem and further the data
size in terms of relevant tokens is limited, boosting
or bagging, which further reduce the data to smaller
subsets of data, are not suitable design options.
2.5.2 Ensemble Combination: Model Assembly
The combination method that combines the results
of the diverse learning methods in the ensemble to
obtain one aggregated consensus result can be achie-
ved through different techniques. The most com-
HEALTHINF 2018 - 11th International Conference on Health Informatics
180

monly used technique is Majority Voting (MV), that
is, selecting as result the class that receives the hig-
hest votes from all the individual learning methods by
simple counting. It can be simple or weighted voting
where base learners are given different weights. In
either case, the average is taken.
Another technique is Stacked Generalization
(Wolpert, 1992) or in short Stacking, which is a lear-
ning over learners method to procure the final result.
Stacking is a meta-learning algorithm where the class
predictions from the base learners are passed as input
data to the meta-algorithm to learn what the correct
output is, given the prediction patterns of the base le-
arner. In our study, we experiment with both Majority
Voting and Stacking techniques as model combiners.
Ultimately, we demonstrate that Stacking method out-
performs Majority Voting and therefore is a promising
strategy to adopt for combining the models into an en-
semble.
2.6 Strategies for Addressing the Class
Imbalance Problem
In biomedical named entity recognition tasks, often
the training datasets used are very skewed, that is,
they suffer from a heavy class imbalance (Nguyen and
Patrick, 2016). Class imbalance occurs when one of
the two classes, usually the class of interest, the po-
sitive class is in stark minority and the negative class
is in majority. The performance of machine learning
methods trained over such class-imbalanced datasets
tend to be greatly affected by such class imbalance.
In particular, this tends to result in the minority class
not being well learned and hence misclassified most
of the time. Class imbalance can influence the per-
formance of the ML method by favoring the majo-
rity negative class. Approaches to deal with class-
imbalanced datasets are described next.
2.6.1 Balancing with Class Weights
One common method is to balance the class weights
within the classifier, thereby giving more importance
(or weight) to the errors of the minority class. Hig-
her class-weight puts more emphasis on the minority
class. That is, it penalizes the model for making clas-
sification mistakes on instances of the minority class
during training. These penalties bias the model to pay
more attention to the minority class.
Usually, in the case of balanced datasets both clas-
ses are given an equal weight of one. In imbalan-
ced datasets however, the class weights can be balan-
ced by performing a grid search with different class
weight combinations to find the optimal class weig-
hts. These weights are then passed to the learning
method to bias the decision making process of the le-
arning method.
2.6.2 Balancing with Class Instances
Another approach to minimize the effect of class im-
balance is to re-sample the original training dataset
to create a new modified training dataset that has a
balanced class distribution. Random over-sampling
and random under-sampling are both common re-
sampling techniques (Chawla, 2009). In both cases,
the objective is to decrease the effect of the highly
skewed class distribution by creating a balance bet-
ween the number of majority and minority class in-
stances. This then enables the classifier to give equal
importance to both classes during the training phase.
However, both techniques have limitations. While
with under-sampling there is a possibility of thro-
wing away important instances, with over-sampling
we tend to increase the size of the training dataset. In
this study, since our training dataset is already large
and high dimensional, we choose to re-sample the da-
taset with the random under-sampling technique.
2.6.3 Balancing with Classifier Ensembles
Yet another approach to deal with class imbalance is
to use ensemble methods to generate a classifier en-
semble that can create a balanced learning environ-
ment for the learning algorithm (Błaszczy
´
nski et al.,
2013). Under-Bagging (Barandela et al., 2003) and
Over-Bagging (Wang and Yao, 2009) are examples of
ensemble techniques, that deal with class imbalance
in the learning phase through a combination of data
re-sampling and bagging approaches, known as “ba-
lanced bagging”.
To the best of our knowledge, with the above exis-
ting methods, the diversity in the ensemble is usually
generated through training one homogeneous learning
algorithm on all balanced subsets of the training data.
The results from the classifier ensemble are aggrega-
ted using the Majority Voting combination method.
In this study, although we will employ the basic idea
of “balanced bagging”, we will also extend it to train
a diverse set of heterogeneous learning algorithms in
parallel.
2.7 Tiered Ensemble Learning System
with Diversity
In this study, to address the two challenges of (1) class
imbalance and (2) the lack of a single best performing
method, we propose a novel integrated approach to
One Size Does Not Fit All: An Ensemble Approach Towards Information Extraction from Adverse Drug Event Narratives
181

Balanced((
Bags(
!"#$%&'(
)%"*+,&"*((
-./''(
)
0121((
)
0123(
)
0124(
)
0125(
)
0321(
)
0323(
)
0324(
)
0325(
)
0421(
)
0423(
)
0424(
)
0425(
)
0521(
)
0523(
)
0524(
)
0525(
)
0621(
)
0623(
)
0624(
)
0625(
21(
23(
24(
25(
Imbalanced((
Data(
Heterogeneous((
Methods(
0
1
2
1(
0
1
2
3(
0
1
2
4(
0
1
2
5(
0
3
2
1(
0
3
2
3(
0
3
2
4(
0
3
2
5(
0
4
2
1(
0
4
2
3(
0
4
2
4(
0
4
2
5(
0
5
2
1(
0
5
2
3(
0
5
2
4(
0
5
2
5(
0
6
2
1(
0
6
2
3(
0
6
2
4(
0
6
2
5(
Classifier((
Ensemble(
Intermediate(
Results(
0(
T
1(
T
2(
T
3(
T
4(
T
5(
7&/,8+9:(
;<"/%9+9:(
$="%(
<"/%9"%'>(
CombinaAon(
Strategy(
Final((
Output(
Figure 4: TELS-D tiered ensemble learning system with diversity.
create a balanced learning environment. This stra-
tegy combines balanced resampling techniques with
an ensemble of heterogeneous classifiers into one
methodology. Our approach called Tiered Ensemble
Learning System with Diversity (TELS-D), effecti-
vely deals with the class imbalance problem in the
data through a balanced under-sampled bagging ap-
proach, while also addressing the limitations of using
a single learning method by training multiple hetero-
geneous learning methods on the under-sampled sub-
sets in parallel.
The imbalance level in a dataset is defined as the
ratio of the number of majority negative class instan-
ces to the number of minority positive class instances
(Eq.1). It indicates how many times the majority class
is greater than the minority class.
Imbalance Level (IM) =
# Negative class tokens
# Positive class tokens
(1)
Based on the imbalance level of a dataset, we create
multiple smaller subsets of the original dataset that
each individually exhibit a balanced class distribution.
That is, each smaller balanced subset takes all of the
available positive class instances while working with
only an equal number of negative class instances, i.e.,
a subset of the available negative class instances. The
purpose here is to learn the features inherent in the po-
sitive class (the class of interest) without getting over-
whelmed by the majority negative class instances and
their typical characteristics. The number of subsets to
form is determined by the imbalance level in the da-
taset. For example, in Fig. 4, the negative class is five
times larger than the positive class. Hence, the origi-
nal unbalanced training dataset (DB) is split into five
smaller balanced subsets henceforth called “balanced
bags” (BB) while ensuring that we do not discard any
instances from either classes, i.e., ∩
5
i=1
BB 6=
/
0 and
∪
5
i=1
BB = DB
For example, if the imbalance level in the dataset
is N, then we create N (N > 1) balanced training sets,
BB. If we have M (M > 1) base learning methods,
we train T = N × M base learners in the first layer of
the ensemble. So, instead of creating an ensemble of
just N diverse models (Sec. 2.5.1-2) or just M diverse
models (Sec. 2.5.1-1), with our proposed TELS-D
strategy we create a collection of T diverse models.
The advantage of TELS-D approach is that we ge-
nerate more diversity in the ensemble while balancing
the class distribution. With more diverse base lear-
ners, each one of the T base classifiers will make dif-
ferent errors on different instances. We then combine
the results from these T diverse base learners to form
an input for the second layer stacking meta-algorithm.
This gives the meta-learner an opportunity to learn the
patterns to predict the correct class - thereby reducing
the total error.
2.8 Evaluation Criteria
We adopt the criteria commonly used for evaluating
classification methods, but now adapt them to apply to
the token-granularity level. That is, we measure both
the Precision and Recall as described below to deter-
mine whether or not the learning models sufficiently
capture the classifications of the positive class.
Precision (P) =
# Correctly predicted positive tokens
# Total predicted positive tokens
(2)
Recall (R) =
# Correctly predicted positive tokens
# Total real positive tokens
(3)
Our goal is to achieve high precision (lesser false
positives) and high recall (more true positives). Thus,
F-measure, defined below, gives a balance between
HEALTHINF 2018 - 11th International Conference on Health Informatics
182

Figure 5: Grid search results for balancing class weight on
target Reason) in i2b2 dataset.
both precision and recall measures, thereby balancing
the accuracy of both positive and negative predictions.
Hence, F-measure is a commonly accepted measure
to evaluate the performance of learning methods.
F-measure (F1) =
2(P x R)
(P + R)
(4)
3 RESULTS
3.1 Experimental Setup
Data Sets. In this study, to build and evaluate our
classification approaches we have used the data set
of annotated patient discharge summaries from i2b2
(Sec.2.1) that has been augmented with ground truth
labels which are needed for supervised machine le-
arning strategies. Holdout test set approach is adop-
ted with a 90/10 split. The i2b2 corpus, the 242 re-
ports used in this study (Table 1) are split accordingly
where 90% of the reports (217 reports) are randomly
selected for training and building our proposed model
and the remaining 10% (25 reports) are used as the
holdout for subsequent testing to evaluate the effecti-
veness of our methods. In this section we discuss our
empirical results on this holdout test set. Additionally,
we have experimented with the 16 FAERS reports as
a second test set (Table 1). Due to lack of ground truth
labels for these FAERS reports, we manually evalua-
ted the results and present a case study as part of our
results discussion.
Parameter Tuning. Base learners such as SVM and
LR must be tuned first and parameters are used to do
so. Therefore, we have used SVM with a linear ker-
nel function and LR with a c-value of 1.0. These va-
lues were the best parameters we obtained after tes-
ting with c-values (0.001, 0.01, 0.1, 1, 10) using 10-
fold cross-validation (Kohavi et al., 1995). The c-
value controls the trade off between model complex-
ity and misclassified instances. We have used deci-
sion tree with best split at each node strategy and gini
to measure the quality of the split (Tan et al., 2006).
For selecting the optimal class weight setting, we per-
formed a systematic grid search with a set of class
weights for each class using 10-fold cross-validation.
The effect of balancing different class weight values
on individual learning methods (SVM/LR/DT) is de-
picted in Fig.5. This experiment shows that for the
three base learners, the precision and recall are ba-
lanced with a higher F-Measure at a class weight
{
C1 : 0.8,C0 : 0.2
}
setting, where C1 denotes the
class reason and C0 denotes the class non-reason.
We thus set the class weight to
{
C1 : 0.8,C0 : 0.2
}
throughout the rest of our experiments where we ba-
lance the class weights within the learning methods.
3.2 Classification with Unbalanced
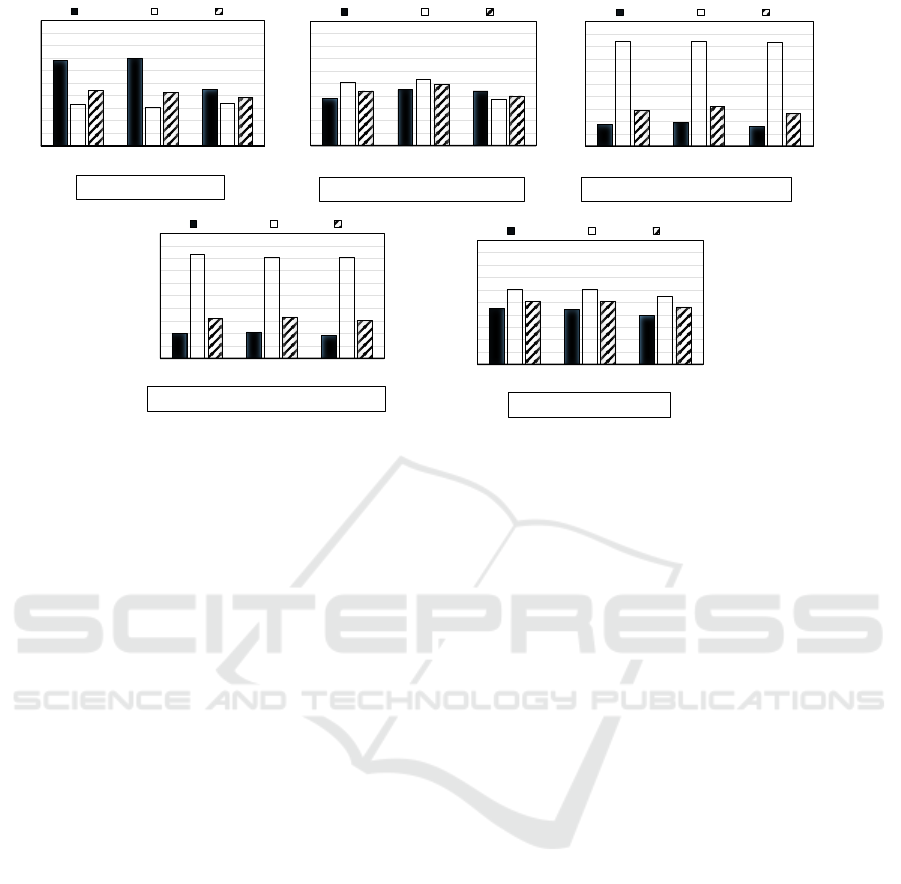
Class Distribution
This experiment is conducted to obtain a baseline
to compare the different approaches explained in
Sec.2.6. The individual base learners are trained on
the original training set (DB) without balancing the
class weights or instances (Fig.6 (a)) to see the effect
of skewed class distribution.
In this experiment, the precision P is much hig-
her than the recall R for all base learners especially
for SVM (P:0.68/ R:0.33) and LR (P:0.70/ R:0.31).
High precision and low recall implies very few to-
kens were predicted as belonging to reason class, but
most of them are correct predictions when compared
against ground truth labels. This is expected due to
the class imbalance, with the majority of the tokens
being non-reason labels in the training phase. Thus
the base classifiers are biased towards the non-reason
class and tend to mis-classify most tokens in the mi-
nority reason class.
3.3 Balancing with Class Weights
This experiment is conducted to evaluate the effecti-
veness of the strategy of balancing class weights to
address the data imbalance problem. The class weight
parameter is set to
{
C1 : 0.8,C0 : 0.2
}
in the indivi-
dual base learners. The base learners are then trained
on the original training set (DB) (Fig.6 (b)).
One Size Does Not Fit All: An Ensemble Approach Towards Information Extraction from Adverse Drug Event Narratives
183
0.68%
0.70%
0.45%
0.33%
0.31%
0.34%
0.44%
0.43%
0.39%
0.00%
0.10%
0.20%
0.30%
0.40%
0.50%
0.60%
0.70%
0.80%
0.90%
1.00%
SVM% LR% DT%
Precision)
Recall)
F1)
F-measure)
Individual)Base)Classifiers)
0.38%
0.45%
0.44%
0.51%
0.53%
0.37%
0.44%
0.49%
0.40%
0.00%
0.10%
0.20%
0.30%
0.40%
0.50%
0.60%
0.70%
0.80%
0.90%
1.00%
SVM% LR% DT%
Precision)
Recall)
F1)
F-measure)
Individual)Base)Classifiers)
0.17%
0.19%
0.16%
0.84% 0.84%
0.83%
0.29%
0.32%
0.26%
0.00%
0.10%
0.20%
0.30%
0.40%
0.50%
0.60%
0.70%
0.80%
0.90%
1.00%
SVM% LR% DT%
Precision)
Recall)
F1)
F-measure)
Individual)Base)Classifiers)
0.20%
0.21%
0.18%
0.83%
0.81% 0.81%
0.32%
0.33%
0.30%
0.00%
0.10%
0.20%
0.30%
0.40%
0.50%
0.60%
0.70%
0.80%
0.90%
1.00%
SVM% LR% DT%
Precision)
Recall)
F1)
F-measure)
Individual)Base)Classifiers)
0.45%
0.44%
0.39%
0.60% 0.60%
0.55%
0.51% 0.51%
0.46%
0.00%
0.10%
0.20%
0.30%
0.40%
0.50%
0.60%
0.70%
0.80%
0.90%
1.00%
SVM% LR% DT%
Precision)
Recall)
F1)
F-measure)
Individual)Base)Classifiers)
a)%Unbalanced%Classes%
b)%Balancing%with%Class%Weights%% c)%Balancing%with%Class%Instances%%%
d)%Balancing%with%Ensemble%Methods%
e)%Balancing%with%TELS-D%%
Figure 6: The precision, recall and F1-score of different classification strategies.
In this experiment, the recall is now higher than
the precision for two base learners, SVM (P:0.38/
R:0.51) and LR (P:0.45/ R:0.53). High recall and low
precision implies many tokens were predicted as be-
longing to reason class. However, most of them are
incorrect predictions when compared against ground
truth labels. This is expected because, in order to deal
with class imbalance during the training phase, we
had set the class weights within the base learners such
that the minority reason class is given more weight.
Hence this tips the classifier learning bias towards the
minority reason class. In contrast to the Unbalan-
ced experimental results (Sec.3.2), this now had led
to more of the majority non-reason class tokens being
misclassified as reason class. The evaluation metrics
of DT (P:0.44/ R:0.37/ F1:0.40) are similar to the un-
balanced experimental results (Sec.3.2).
3.4 Balancing with Class Instances
The next experiment evaluates the effect of balancing
class instances to address the class imbalance pro-
blem. Balancing class instances is achieved by perfor-
ming random under-sampling on the original training
dataset (DB) to create a single balanced subset of the
training data to be utilized for training. The resulting
balanced subset now has an equal number of positive
reason and negative non-reason class instances (Fig.6
(c)).
In this experiment, the recall is much higher than
precision for all base learners, SVM (P:0.17/ R:0.84),
LR (P:0.19/ R:0.84) and DT (P:0.16/ R:0.83). In fact,
the precision is rather low. This indicates that most of
the tokens were predicted as belonging to the reason
class, when in actuality a majority of them belongs to
the non-reason class. This also explains the very high
recall, where most of the ground truth labels were
also included in the total predictions. This can be ex-
plained by the fact that during under-sampling only a
random subset of negative class non-reason instances
were included in the balanced subset. Hence we dis-
carded many potentially useful instances that are im-
portant for learning the reason class. In this scenario,
the base learners cannot learn the predominant cha-
racteristics of the negative class well and hence tend
to mis-classify those instances more often.
3.5 Balancing with Classifier Ensembles
This experiment evaluates the effect of balancing with
ensemble of homogeneous classifiers. Balancing with
Ensemble of Homogeneous Classifiers is achieved
by performing Under-Bagging strategy on the origi-
nal training dataset (DB) to create multiple under-
sampled subsets of the training data (Sec. 2.6.3).
Then we train each base learner on all of these sub-
sets. Lastly, we combine them with Majority Voting
(Fig.6 (d)).
In this experiment, the recall is much hig-
her than the precision for all base learners, SVM
(P:0.20/ R:0.83), LR (P:0.21/ R:0.81) and DT (P:0.18/
R:0.81). These results are similar to the experimental
results of Balancing with Class Instances (Sec.3.4).
Although, both approaches are similar in the crea-
tion of a balanced subset, this current approach uses
multiple balanced subsets to counter the limitations of
using a single balanced subset (i.e. eliminating poten-
tially important negative class instances). However,
HEALTHINF 2018 - 11th International Conference on Health Informatics
184
0.44$ 0.44$
0.29$
0.32$
0.51$
0.43$
0.49$
0.32$
0.33$
0.51$
0.39$
0.40$
0.26$
0.30$
0.46$
0.00$
0.05$
0.10$
0.15$
0.20$
0.25$
0.30$
0.35$
0.40$
0.45$
0.50$
0.55$
0.60$
Un-Balanced$Class$
Distribu<on$
Balancing$w$Class$
Weights$
Balancing$w$Class$
Instances$
Balancing$w$
Classifier$
Ensembles$
Balancing$w$$$$$$$$$$$$$$$$$$$$$$$$$$$$$$$$$$$$
TELS-D$
SVM$
LR$
DT$
Classifica(on+Strategies+for+Class+Imbalance+
F-Measure$
Figure 7: Comparison of classification strategies for class
imbalance - F-measures of base classifiers.
the Under-Bagging approach uses majority voting to
aggregate the results obtained from training the base
classifiers on these subsets. We see (Fig.6 (c)) that the
precision on a single subset is very low. So even if we
take a majority vote of N such classifiers whose indi-
vidual base results are erroneous, the final prediction
tends to be also incorrect.
3.6 Balancing with TELS-D
Our proposed approach, TELS-D is a multi-layer fra-
mework (Sec. 2.7). The first layer in TELS-D creates
a balanced learning environment to handle class im-
balance in the training dataset.
This experiment evaluates the first layer in TELS-
D. Balancing is achieved by creating multiple balan-
ced subsets (BB) of the original training data (DB) ba-
sed on the imbalanced level (IM) in the training set.
We train each base learner on the balanced subsets
(BB) and combine them with Stacking, using anot-
her meta-algorithm (Logistic Regression). In contrast
to Under-Bagging which uses simple majority voting,
TELS-D employs stacking method to combine the re-
sults from the base learners and make the final pre-
dictions (Fig.6 (e)).
In this experiment, the recall is a little higher than
precision for all base learners, SVM (P:0.45/ R:0.60),
LR (P:0.44/ R:0.60) and, DT (P:0.39/ R:0.55). That
is, although we have predicted many of the tokens
correctly, some of the class predictions were incor-
rect when compared against ground truth labels. This
small learning bias towards the minority reason class
is expected because, during the training phase, we
give priority to learning the minority reason class well
by training on multiple subsets that have the same mi-
nority instances.
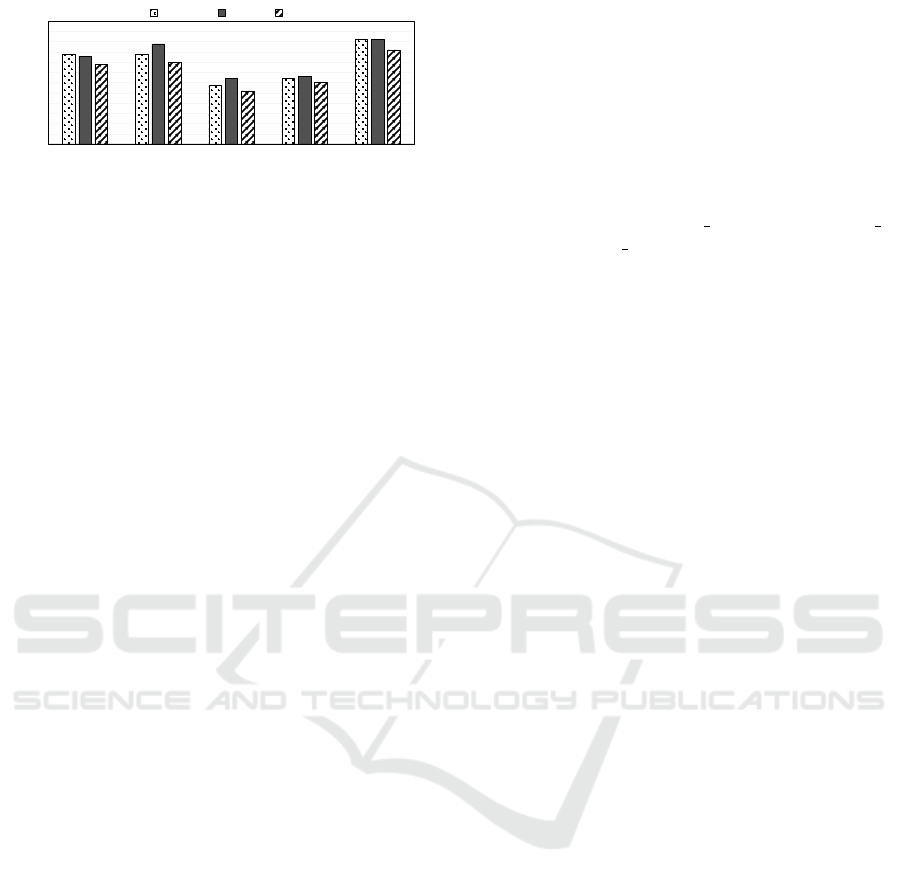
3.7 Comparing Classification Strategies
for Class Imbalance
To compare our experimental results of different ap-
proaches for dealing with class imbalance, we evalu-
ate their performances on each individual base lear-
ners using the F-Measure metric. F-Measure gives a
weighted average of the precision and recall scores.
An improvement is the F-measure indicates an equi-
librium point where we increase the number of cor-
rect class predictions thereby decreasing the number
of incorrect class predictions. Fig. 7 shows that our
proposed TELS-D approach is effective in solving the
class imbalance problem with higher F-Measures on
all three base learners (SVM F-Measure:0.51/ LR F-
Measure:0.51/ DT F-Measure:0.46) compared to ot-
her approaches that deal with class imbalance.
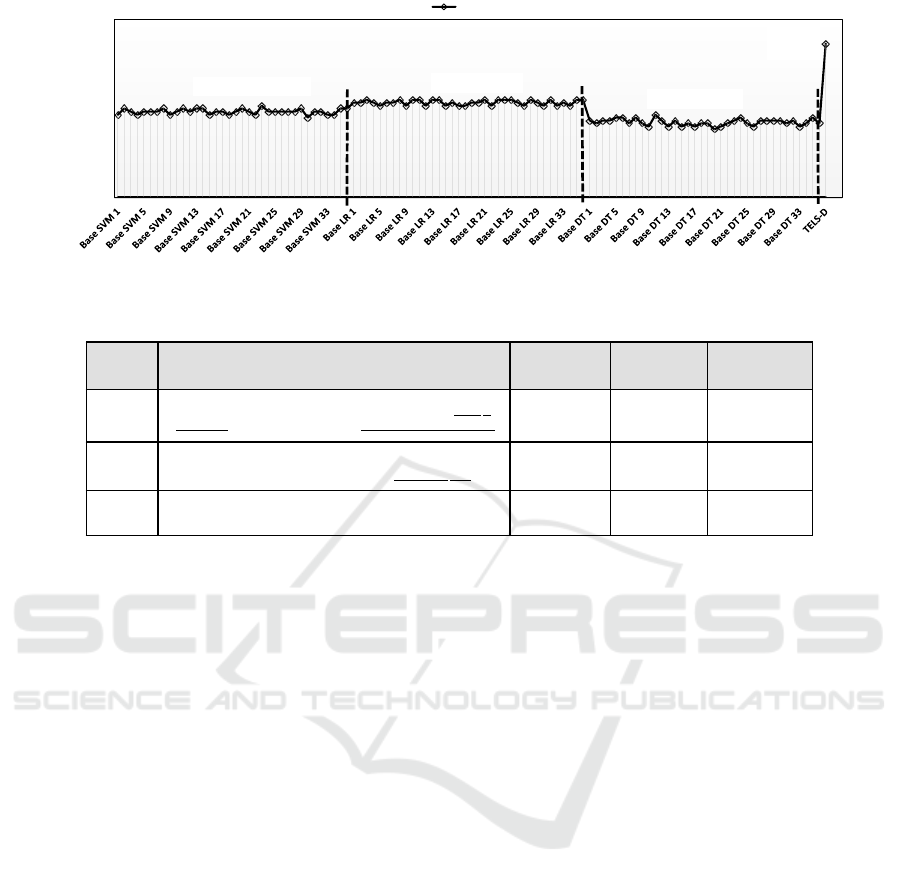
3.8 Ensemble Learning with TELS-D
The second layer in TELS-D is designed to create and
combine an ensemble of heterogeneous classifiers to
improve the accuracy over the individual base learners
(Sec. 2.7). This experiment evaluates the second layer
of TELS-D built on the output from the first layer. The
predictions of the three base learners trained over all
balanced subsets in the first layer are combined with
Stacking using a meta-algorithm, in our case a simple
linear algorithm like Logistic Regression (LR), in the
second layer.
Fig. 8 shows F-Measure of: 1) Individual base
classifiers generated by training the three base lear-
ners on all balanced subsets, 2) Ensemble combined
with majority voting (for comparison only) and, 3)
Ensemble combined with Stacking. The F-measures
of individual base classifiers were ranging from 0.28-
0.33, Ensemble with Majority voting is 0.22 whereas
the F-measure of the ensemble with stacking is 0.52.
This experiment demonstrates the power of an en-
semble learning system with a learning-over-learners
combiner called meta-algorithm in the final step. The
meta-algorithm learns from the errors generated by
the base classifiers to output the correct result. Majo-
rity voting on the other hand is under performing due
to the fact that, with simple counting of votes, the er-
rors of the base classifiers only add up and thus make
the final result more erroneous.
We have compared our results with an existing
study (Doan and Xu, 2010) conducted on the same
i2b2 test dataset. (Doan and Xu, 2010) demonstra-
ted with MedEx only and SVM-based NER including
MedEx. The results showed that for recognizing the
reason entity from the narratives, the rule-based Me-
dEx system achieved a F-measure of 0.43 while the
SVM combined with MedEx achieved 0.48. Our re-
sults from TELS-D approach show an improvement
over both MedEx and SVM including MedEx with
the F-measure of 0.52.
One Size Does Not Fit All: An Ensemble Approach Towards Information Extraction from Adverse Drug Event Narratives
185
Base%SVM1%-%SVM36%
Base%LR1%-%LR36%
Base%DT1%-%DT36%
TELS-D%
F1=%0.52%
0.00%
0.10%
0.20%
0.30%
0.40%
0.50%
0.60%
F1#
F-Measure%
Figure 8: Evaluation of tiered ensemble learning system with diversity (TELS-D).
Table 2: FAERS Examples of Reason class labels predicted by TELS-D.
Example Sentence from the FAERS Narrative
True Positive
(TP)
False Positive
(FP)
False Negative
(FN)
1)
The patient was treated with canagliflozin for type 2
diabetes and domperidone for diabetic gastroparesis
type, diabetes,
diabetic,
gastroparesis
! 2
2)
The patient had previously experienced allergy when
taking mycins (antibacterials for systemic use)
systemic ! use
3)
Concurrent conditions abdominal pain, diabetic paresis.
!
abdominal,
pain, diabetic
!
3.9 Analysis of TELS-D Results on
FAERS Reports
Due to lack of ground truth labels for FAERS reports,
we manually reviewed and evaluated the TELS-D re-
sults on few of the 16 FAERS reports. An analysis
of errors on one of the FAERS narrative is discussed
below (See Table 2).
• True Positives: True Positives (TP) are the cor-
rectly predicted tokens. In the Table 2, we can ob-
serve that for examples 1 and 2 all the tokens labeled
as reason class have been accurately predicted as true
positives by our TELS-D. Most of the ground truth
labeled words in these sentences are purely medical
text and follow a certain sentence structure.
• False Positives: False positives (FP), i.e., incor-
rectly predicted as reason class, mostly occurred
when the token was not associated with a medication.
For instance, example 3 shows that although the in-
correctly predicted token is all medical text, it was
not associated with a medication name in the same
sentence. Hence it cannot be an indication for taking
a medication and is not predicted as reason. Cases
such as these are very difficult to classify and indicate
a need for additional features to learn such patterns in
the text.
• False Negatives: Our evaluation showed that false
negatives, i.e., incorrectly predicted as non-reason
class, occurred primarily due to the mixture of me-
dical and non-medical words. Most of the time, we
have noticed that these false negative tokens are em-
bedded or were a part of the true positive tokens. For
instance, in examples 1 and 2, the words “2”, “use”
are all commonly used regular text.
4 DISCUSSION
Lack of Annotated FAERS Dataset. First, FAERS
narratives cannot be published without data redaction
because of privacy concerns. Redaction of these re-
ports requires a huge amount of cautious efforts to
make sure no privacy threatening information remains
in the publishable text. Since the redaction process
requires perfect recall with utmost precision, it is
almost impossible to be accomplished automatically
without significant manual intervention. Therefore,
creating a large corpus of redacted FAERS narratives
is challenging in itself. Second, annotating FAERS
narrative requires deep domain knowledge and revie-
wing experiences. Deployable supervised machine le-
arning models used for such task must be trained on
larger datasets annotated by FDA’s own safety revie-
wers whose annotating strategy reflects the reviewing
guidelines. However, due to limited resources, anno-
tating a large set of FAERS narratives is not trivial
as it requires extra effort and time in addition to the
HEALTHINF 2018 - 11th International Conference on Health Informatics
186

routine drug review tasks. Given the above challen-
ges, there are no publishable FAERS reports annota-
ted by FDA that can be used in this study for training
and testing purposes. Therefore, to prove the concept
and for the reproducibility of this study, we trained
our model and evaluated our methodology using the
public benchmark dataset (i2b2 2009 discharge sum-
maries). In addition, we tested the trained model on a
few redacted FAERS narratives that have been anno-
tated. Since discharge summaries do not necessarily
share the same vocabulary as the FAERS narratives,
we expect this switch in data sets to be reflected in
the results as well.
Practical Application of this Study for FDA. Au-
tomatically identifying high value information from
the biomedical text has been recognized by FDA as
one of the important steps in its regulatory and su-
pervisory tasks. FDA has been partnering with re-
search institutes and technology companies to deve-
lop text mining and natural language processing tools
for various types of biomedical text collected by FDA
such as vaccine ADR reports (VAERS), FAERS re-
ports, and others. Due to the different nature of these
texts, the tools and methodologies are highly custo-
mized to work with a particular text type. Moreover,
among these text types, FAERS narratives have rela-
tively complex structure in terms of size, vocabulary
and style of writing. To cope with this complexity, we
propose a machine learning framework that can com-
bine some of these internally available existing tools
to extract information from FAERS narratives in an
ensemble fashion. These extracted results can be furt-
her utilized by advanced data mining or visualization
techniques to enhance the drug review process.
5 CONCLUSIONS
This paper describes a novel approach called Tiered
Ensemble Learning System with Diversity (TELS-D)
for biomedical NER from Adverse Event Reports.
Our proposed approach uses an ensemble of diverse
heterogeneous classification methods to recognize na-
med entities in the text while also dealing with the
critical problem of skewed class distribution of the
named entities in the training datasets. Our results
are promising and indicate that, in the context of bi-
nary classification an ensemble approach would be a
better choice for NER especially for class imbalanced
datasets.
REFERENCES
Alpaydin, E. (2014). Introduction to machine learning.
MIT press.
Aronson, A. R. (2001). Effective mapping of biomedical
text to the umls metathesaurus: the metamap program.
In Proceedings of the AMIA Symposium, page 17.
AMIA.
Barandela, R., Valdovinos, R. M., and S
´
anchez, J. S. (2003).
New applications of ensembles of classifiers. Pattern
Analysis & Applications, 6(3):245–256.
Bird, S. et al. (2009). Natural language processing with Py-
thon: analyzing text with the natural language toolkit.
” O’Reilly Media, Inc.”.
Bishop, C. M. (2006). Pattern recognition and machine le-
arning. springer.
Błaszczy
´
nski, J., Stefanowski, J., and Idkowiak, Ł. (2013).
Extending bagging for imbalanced data. In Procee-
dings of the 8th International Conference on Com-
puter Recognition Systems CORES 2013, pages 269–
278. Springer.
Breiman, L. (1996). Bagging predictors. Machine learning,
24(2):123–140.
Charniak, E. and Johnson, M. (2005). Coarse-to-fine n-best
parsing and maxent discriminative reranking. In Pro-
ceedings of the 43rd Annual Meeting on ACL, pages
173–180. ACL.
Chawla, N. V. (2009). Data mining for imbalanced data-
sets: An overview. In Data mining and knowledge
discovery handbook, pages 875–886. Springer.
Doan, S. and Xu, H. (2010). Recognizing medication re-
lated entities in hospital discharge summaries using
support vector machine. In Proceedings of the 23rd
International Conference on Computational Linguis-
tics: Posters, pages 259–266. ACL.
FDA (2016). FAERS (FDA adverse event reporting sy-
stem).
Feng, X., Cai, A., Dong, K., Chaing, W., Feng, M., Bhu-
tada, N. S., Inciardi, J., and Woldemariam, T. (2013).
Assessing pancreatic cancer risk associated with di-
peptidyl peptidase 4 inhibitors: Data mining of fda ad-
verse event reporting system (faers). Journal of Phar-
macovigilance, pages 1–7.
Ferrucci, D. and Lally, A. (2004). Uima: an architectu-
ral approach to unstructured information processing
in the corporate research environment. Natural Lan-
guage Engineering, 10(3-4):327–348.
Friedman, C., Alderson, P. O., Austin, J. H., Cimino,
J. J., and Johnson, S. B. (1994). A general natural-
language text processor for clinical radiology. JAMIA,
1(2):161–174.
Galar, M., Fernandez, A., Barrenechea, E., Bustince, H.,
and Herrera, F. (2012). A review on ensembles for
the class imbalance problem: bagging-, boosting-, and
hybrid-based approaches. IEEE Transactions on Sys-
tems, Man, and Cybernetics, Part C (Applications and
Reviews), 42(4):463–484.
Ghiasvand, O. (2014). Disease name extraction from clini-
cal text using conditional random fields. PhD thesis,
The University of Wisconsin-Milwaukee.
One Size Does Not Fit All: An Ensemble Approach Towards Information Extraction from Adverse Drug Event Narratives
187

Halgrim, S. R., Xia, F., Solti, I., Cadag, E., and Uzuner,
¨
O.
(2011). A cascade of classifiers for extracting medica-
tion information from discharge summaries. Journal
of biomedical semantics, 2(3):S2.
Harpaz, R., Callahan, A., Tamang, S., Low, Y., Odgers, D.,
Finlayson, S., Jung, K., LePendu, P., and Shah, N. H.
(2014). Text mining for adverse drug events: the pro-
mise, challenges, and state of the art. Drug safety,
37(10):777–790.
Jagannatha, A. N. and Yu, H. (2016). Bidirectional rnn for
medical event detection in electronic health records.
In Proceedings of the conference. ACL. North Ameri-
can Chapter. Meeting, volume 2016, page 473. NIH
Public Access.
Japkowicz, N. and Stephen, S. (2002). The class imbalance
problem: A systematic study. Intelligent data analy-
sis, 6(5):429–449.
Kohavi, R. et al. (1995). A study of cross-validation and
bootstrap for accuracy estimation and model selection.
In Ijcai, volume 14, pages 1137–1145. Stanford, CA.
Kuhn, M., Letunic, I., Jensen, L. J., and Bork, P. (2015).
The sider database of drugs and side effects. Nucleic
acids research, 44(D1):D1075–D1079.
Lazarou, J., Pomeranz, B. H., and Corey, P. N. (1998). In-
cidence of adverse drug reactions in hospitalized pa-
tients: a meta-analysis of prospective studies. Jama,
279(15):1200–1205.
Longadge, R. and Dongre, S. (2013). Class imbalance
problem in data mining review. arXiv preprint
arXiv:1305.1707.
Nguyen, H. and Patrick, J. (2016). Text mining in clinical
domain: Dealing with noise. In KDD, pages 549–558.
Polikar, R. (2009). Ensemble learning. Scholarpedia,
4(1):2776. revision #91224.
Ramesh, B. P., Belknap, S. M., Li, Z., Frid, N., West, D. P.,
and Yu, H. (2014). Automatically recognizing medi-
cation and adverse event information from food and
drug administrations adverse event reporting system
narratives. JMIR medical informatics, 2(1):e10.
Sakaeda, T., Tamon, A., Kadoyama, K., and Okuno, Y.
(2013). Data mining of the public version of the fda
adverse event reporting system. International journal
of medical sciences, 10(7):796.
Savova, G. K., Masanz, J. J., Ogren, P. V., Zheng, J., Sohn,
S., Kipper-Schuler, K. C., and Chute, C. G. (2010).
Mayo clinical text analysis and knowledge extraction
system (ctakes): architecture, component evaluation
and applications. JAMIA, 17(5).
Schapire, R. E. (1990). The strength of weak learnability.
Machine learning, 5(2):197–227.
Tan, P.-N. et al. (2006). Introduction to data mining. Pear-
son Education India.
Uzuner,
¨
O., Solti, I., and Cadag, E. (2010a). Extracting
medication information from clinical text. JAMIA,
17(5):514–518.
Uzuner,
¨
O., Solti, I., Xia, F., and Cadag, E. (2010b). Com-
munity annotation experiment for ground truth ge-
neration for the i2b2 medication challenge. JAMIA,
17(5):519–523.
Uzuner,
¨
O., Zhang, X., and Sibanda, T. (2009). Machine
learning and rule-based approaches to assertion clas-
sification. JAMIA, 16(1):109–115.
Wang, S. and Yao, X. (2009). Diversity analysis on imba-
lanced data sets by using ensemble models. In Pro-
ceedings of the IEEE Symposium on Computational
Intelligence and Data Mining, CIDM, pages 324–331.
Wilson, A. M., Thabane, L., and Holbrook, A. (2004). Ap-
plication of data mining techniques in pharmacovigi-
lance. BJCP, 57(2):127–134.
Wolpert, D. H. (1992). Stacked generalization. Neural net-
works, 5(2):241–259.
Xu, H., Stenner, S. P., Doan, S., Johnson, K. B., Waitman,
L. R., and Denny, J. C. (2010). Medex: a medication
information extraction system for clinical narratives.
JAMIA, 17(1):19–24.
HEALTHINF 2018 - 11th International Conference on Health Informatics
188
