
Protein Disorder Prediction using Jumping Motifs from Torsion Angles
Dynamics in Ramachandran Plots
Jonny Alexander-Uribe
1
, Juli
´
an D. Arias-Londo
˜
no
1
and Alexandre Perera-Lluna
2
1
Department of Systems Engineering and Computer Science, Universidad de Antioquia,
Calle 67 No. 53 - 108, 050010, Medell
´
ın, Colombia
2
Research Center for Biomedical Engineering, ESAII, Universitat Polit
`
ecnica de Catalunya,
Pau Gargallo 5, 08028, Barcelona, Spain
Keywords:
Intrinsically Disordered Proteins, Intrinsically Disordered Regions, Dihedral Torsion Angles, Ramachandran
Plot, Conditional Random Fields.
Abstract:
Disordered proteins are functional proteins that do not fold in a fixed 3D structure. The order/disorder pre-
diction in protein sequences is an important task given the biological roles of disordered proteins. In the last
decade many computational based methods have been proposed for the disorder identification but currently
the most accurate strategies depend on the sequence alignment of large databases of proteins, making the
methods slow and hard to apply on proteome-wide analysis. In this paper is proposed an innovative approach
for linking the amino acid sequences with transition tendencies in their dihedral torsion angles. The aim is to
characterize the dynamical angle variations along the protein chain, as a way of measuring the flexibility of the
amino acids and its connection with the disorder state. The features are estimated from empirical propensities
computed from Ramachandran Plots. The classification is performed using structural learning in the form of
CRF (Conditional Random Fields). The performance is evaluated in terms of AUC (Area Under the ROC
Curve), and three suitable performance metrics for unbalanced classification problems. The results show that
the proposed method outperforms the most referenced alignment-free predictors and its performance is also
competitive with the slower and mature alignment-based methods.
1 INTRODUCTION
For many years it was thought that proteins had to
fold in a fixed 3D structure to accomplish their bio-
logical functions. When some experiments showed
the existence of proteins in a chaotic state, which re-
mained without folding, they were initially consid-
ered as anomalies or errors in the experiment (De-
Forte and Uversky, 2016). But as these weird proteins
accumulated, they could no longer be ignored and the
paradigm of sequence → 3D structure → function,
had to be reevaluated for accepting that some proteins
are biologically relevant and must persist in a flexible
configuration (DeForte and Uversky, 2016).
These proteins are now called disordered proteins:
biologically active proteins, which do not have a spe-
cific 3D-structure under normal physiological con-
ditions. When the complete protein remains with-
out a fixed tertiary structure and persist in a flexible
state, the term intrinsically disordered protein (IDP)
is used. In contrast, when a protein is mostly struc-
tured but displays some regions of disorder, it is said
to have intrinsically disordered protein regions (ID-
PRs) (DeForte and Uversky, 2016). Prevalence of dis-
order in nature is global, all organism have IDPs or
IDPR, being estimated that 30% of eukaryotic pro-
teins have long disordered regions (gretear than 30
residues) (Ward et al., 2004). This fact reinforces the
idea that IDPs are part of a clever mechanism for ac-
complishing complex functions that completely fold
proteins could not do.
The biological importance of IDPs is high, they
participate in essential celular processes such as
molecular regulation, transport and signaling. Addi-
tionally, they were found to be associated with hu-
man diseases including cancer, diabetes, cardiovascu-
lar affection, amyloidoses and neurodegenerative dis-
eases (Uversky et al., 2008). Because of that, in recent
years the discovery and characterization of disordered
proteins has become in one of the fastest growing ar-
eas in protein science (He et al., 2009). Neverthe-
less, experimental determination of IDPs and IDPRs
38
Alexander-Uribe, J., Arias-Londoño, J. and Perera-Lluna, A.
Protein Disorder Prediction using Jumping Motifs from Torsion Angles Dynamics in Ramachandran Plots.
DOI: 10.5220/0006647900380048
In Proceedings of the 11th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2018) - Volume 3: BIOINFORMATICS, pages 38-48
ISBN: 978-989-758-280-6
Copyright © 2018 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

poses a costly and complex challenge, requiring both,
a lot of time and an extensive human expertise (He
et al., 2009). Computational methods have become
a valuable alternative to process the large amount of
proteins sequences available and infer their disorder
states. Although several predictors of IDPs have been
proposed in the past, there is still need of faster and
accurate methods for protein disorder identification
(Peng et al., 2015),(Varadi et al., 2015).
The use of Multiple Sequence Alignment (MSA)
algorithms is the main distinguishing characteris-
tic of the current computational methods for detect-
ing disorder. Predictors using MSA, commonly ap-
ply several iterations of PSI-BLAST (Altschul et al.,
1990)(Altschul et al., 1997) for identifying proteins
homologues in known databases. This preliminar
phase, allows the creation of tuned Position Score
Matrices (PSSM). The PSSM can capture the statis-
tical variations of every amino acid on targeted pro-
teins, and are used as inputs for the disorder pre-
dictors, improving their performance in comparison
with the use of only the raw protein sequences. Al-
though the sequence alignment can offer an advan-
tage in the accuracy of the methods, it also imposes
a set of methodological and practical issues. One of
them is the computational cost, which becomes rele-
vant when the method is used on large scale proteome
analysis (thousands to millions of proteins). A sec-
ond and more relevant drawback, is the implicit as-
sumption that the proteins under evaluation have ho-
mologous sequences into the used databases. Some
of the methods that take advantage of the MSA algo-
rithms for identifying disorder include PONDR (Xue
et al., 2010), DISOPRED (Jones and Cozzetto, 2014),
NORSnet (Schlessinger et al., 2007) and SPINE-D
(Zhang et al., 2012).
In contrast, methods that avoid sequence align-
ment can reach more modest classification results on
known datasets, but can be applied comparatively
faster on huge databases of unlabeled proteins (De-
Forte and Uversky, 2016), and more importantly, they
do not make assumptions about the existence of ho-
mologues proteins.
Among the most used alignment-free methods for
protein disorder prediction are IUPRED and Espritz
(Dosztnyi et al., 2005), (Walsh et al., 2012). IUPRED
uses the amino acid pair interaction energy estimated
using only the amino acid compositions, to create ma-
trices of potentials between amino acids. The au-
thors concluded that when a sequence contains few
hydrophobic residues, the composition-based mutual
interaction energy will be small, indicating the lack
of potential for folding. In IUPRED the scoring ma-
trices were adjusted using a Support Vector Machine
(SVM) (Cortes and Vapnik, 1995) and independent
models were created for short and long disorder re-
gions. IUPRED is computationally fast and have
been used in proteome-wide analyses (Oates et al.,
2013) (Potenza et al., 2015). The systems that use
predictors ensembles (metapredictors) recurrently in-
cluded IUPRED as a component (Bulashevska and
Eils, 2008) (Lieutaud et al., 2008), and in many
works where new predictors are proposed, IUPRED
is used as a baseline for comparison purposes (He
et al., 2009)(Deng et al., 2012). On the other hand,
Espritz is based on a Bidirectional Recursive Neu-
ral Network whose inputs are 5 scales obtained from
the clustering of AAindex properties (Kawashima and
Kanehisa, 2000), and a one-hot enconding vector of
length 20, which identify the amino acid being mod-
eled/evaluated at a time. It means that, given an amino
acid, this property vector will have a value 1 for only
one position, and 0s for the 19 other positions. Espritz
is also a fast predictor used in similar scenarios than
IUPRED and therefore well suitable for performance
comparison.
A common strategy for improving the perfor-
mance in disorder vs order classification, is to com-
bine the outputs of several individual predictors, cre-
ating a metapredictor. This combination is often ap-
plied at the residue level where the probability out-
puts from different methods are fusioned into a new
classification phase. Examples of metapredictors are:
MetaPdDOS (Bulashevska and Eils, 2008), MFDp
(Mizianty et al., 2010), MeDor (Lieutaud et al., 2008)
and Metadisorder (MD) (Kozlowski and Bujnicki,
2012).
In spite of the multiple efforts for introducing
more elevorated classification strategies, the perfor-
mance of disorder predictors still has room for im-
provement. A valid approach to increase the protein
disorder classification accuracy, is to create new fea-
tures capable of extracting relevant information from
the sequence, that can be related to the folded or un-
folded state of proteins. A promisory idea is to link
the protein sequence with the dihedral torsion an-
gles of the amino acid chain. This could be rele-
vant because these angles contain information about
restrictions, allowed values and tendencies associated
to the final structure of proteins (Hollingsworth and
Karplus, 2010). This idea was explored in (Baruah
et al., 2015), where the dihedral angles were used
with the aim of estimating the conformational entropy
of IDP, IDPR, and completely ordered proteins. The
proposed metric was found to be a potential mea-
sure for the discrimination of complete disordered vs
complete ordered proteins. In (Uribe et al., 2017) a
set of information theory measures derived from tor-
Protein Disorder Prediction using Jumping Motifs from Torsion Angles Dynamics in Ramachandran Plots
39

sion angles extracted from Ramachandran plots (RP)
were also found to be relevant in the detection of IDP
and IDPR, when they were combined with other well-
established features in the state of the art for disorder
prediction.
In this work, an innovative characterization that
links the inferred torsion angles dynamic along the
chain with the disorder state, is proposed. The
strategy uses the RP distributions for quantifying
the amino acid tendency to jump between confor-
mational regions, transforming the idea proposed in
(Hollingsworth et al., 2012), in a practical tool for
characterizing proteins. The results will show that
using only the information from RPs, it is possible
to design a valuable feature extraction phase, which
once incorporated in a classifier, is able to achieve
similar performance metrics than MSA based meth-
ods, but with a methodology that can be efficiently
computed on millions of proteins. This characteri-
zation called the jumping Motifs, was used here for
the creation of a disorder predictor called jMotCRiF.
The proposed predictor was build using a structural
learning scheme based on Conditional Random Fields
(CRFs) (Lafferty et al., 2001). CRFs are discrimina-
tive non-parametric models able to capture the cor-
relation amongst neighboring labels in a sequence,
therefore they are well suitable for the annotation of
amino acids as ordered/disorder (Uribe et al., 2017).
CRFs were first used in the identification of disor-
dered residues on (Wang and Sauer, 2008), but there
authors used a completely different characterization
based on conventional chemical properties.
The rest of the papers is organized as follows:
section 2 presents the characterization proposed and
describes the learning strategies. It also refers the
dataset used and the applied validation methodology.
Section 3 presents the results obtained and finally sec-
tion 4 includes some conclusions extracted from the
work.
2 MATERIAL AND METHODS
2.1 Characterization
Proteins are linear chains of connected amino acids
that can have hundreds to thousands of elements.
Neighbor amino acids, in order to avoid atomic
clashes, must limit their possible configurations. In
the backbone structure of an amino acid, there are
mainly two angles of turn for every residue: the tor-
sion angles known as φ and ψ. For illustrating this
Figure 1 shows a small stick and ball diagram of a
short subchain of amino acids where the torsion an-
gles are depicted. In this sense, the RPs are 2D repre-
sentations of the variation of φ and ψ angles on known
proteins. The 20 amino acids have different prefer-
ence in the φ and ψ space, because differences in
the three-dimensional structure of the residues, con-
fer different ranges of flexibility. For example, the
residue in Glycine is just a single atom of hydrogen
giving the molecule the highest flexibility and the pos-
sibility of exploring the bigest φ and ψ space. In con-
trast, Proline has a backbone covalent link, that im-
poses strong rigidity on the molecule, reducing the
possible φ and ψ valid angles to minimum. Other
amino acids have intermediate constrains that allow
them to explore different zones in the RPs. Figure 2
shows the RPs of Proline and Glycine, along with two
other representative amino acids.
Ψi
Φi
+1
Ψi
+1
VAL ALA ASP GLY
Φi
Figure 1: Ball diagram of small subchain with residues
Valine (VAL), Alanine (ALa), Aspartic Acid (ASP) and
Glycine (GLY). Φ and ψ torsion angles are shown around α
carbons of ASP and GLY.
2.1.1 The Jumping Motifs
An ideal procedure for the identification of the protein
structure conformation, would take the amino acid
sequence and will predict the torsion angle dynamic
along the chain. Such method does not exists yet but
indirect measures related with this task, can be dis-
cerned using the amino acid propensities computed
from the thousands of known folded proteins.
In (Kalmankar et al., 2014), using proteins from
PDB (Berman et al., 2007), the authors constructed
amino acid propensities for 14 differentiated regions
on the RPs. Many of these zones have direct con-
nection with the secondary structures found in folded
proteins but the sparsely populated zones, in appar-
ently disallowed regions, are also considered. Overall
these propensities capture relevant torsion angle con-
figurations and some preferences that amino acids fol-
low, quantifying the tendency of sets of amino acids
to inhabit particular RP zones.
In Figure 3 the 14 zones are depicted, along with
some of the amino acids more frequently found inside
BIOINFORMATICS 2018 - 9th International Conference on Bioinformatics Models, Methods and Algorithms
40
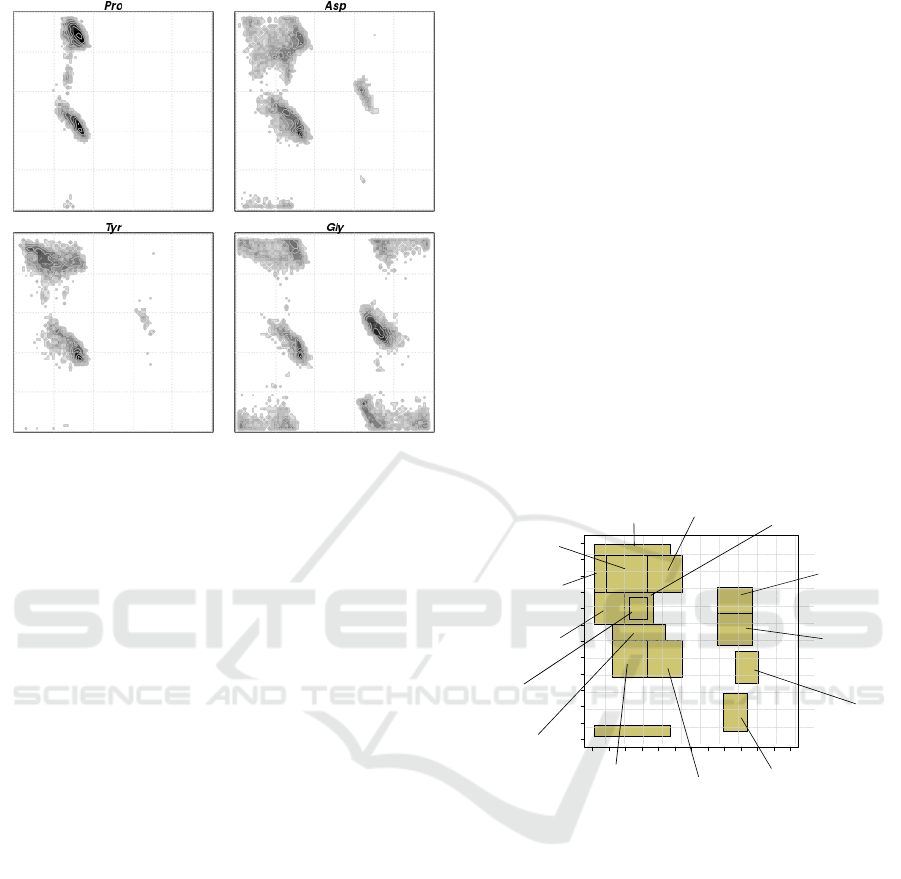
Figure 2: Ramachandran Plots of some amino acids, Pro-
line, Aspartic Acid, Tyrosine and Glycine. φ is along x-axis
and ψ is along y-axis.
these regions. Region number 8 is the most crowded
zone with around 40% of the amino acids analyzed,
inhabiting there. The primarily reason for this, is that
the most common secondary structure, the α helix,
belongs to this region. Zones 1, 2 and 9 together,
contribute with another 40% of the observations. In
region 1, resides the β sheet, the second most com-
mon secondary structure. Poliproline II is in region
number 2 and the inverted α helix is in region 11.
A remarkable contribution of the division made by
(Kalmankar et al., 2014), is that poor favored regions
are also included. For example region 12 contains the
less common structure called γ turn; inverted γ turns
are in region 5 and the type II β turns are in zone num-
ber 13. To consider the low inhabited regions, allows
to capture a more complete dynamic characterization,
covering the entire set of possible torsion angle con-
figurations.
As stated before, amino acids have different ten-
dencies to inhabit the RP regions. Propensities of
some of the residues are depicted in Figure 3. This
preferences are not deterministic, instead must be
treated as stochastic in nature, showing only the statis-
tically most common states. It is also true, that some
amino acids are rarely found in certain regions and
its appearance constitute a unexpected event. These
complementary and opposed dispositions, are con-
cretely quantified in Table 1, where the tendencies of
some amino acids to reside in the different regions
and the tendencies of some of them for avoiding the
zones, are specified numerically.
Given a short protein sequence, is possible to in-
spect their residues and identify which of the RP re-
gions are “activated” by the amino acids in consid-
eration. This activation could also be quantified by
the propensities associated to the residues, in such
way that if many amino acids coincide in the activated
zone its activation intensity would be higher.
Although the resulting activation patterns could be
useful, it would be better to capture, not only the pre-
ferred regions by the sequence, but also its transition
preferences. That is to say, a quantification of the
dynamic change between the RPs regions, could be
of major interest, because of the fact that disordered
amino acids are presumably changing continuously
their torsion angles states, not resting in any partic-
ular spot for long but restlessly jumping between re-
gions. In this way, for identifying the IDPs, an in-
direct measure about the jumping dynamic between
regions, would be useful for inferring the transition
preferences and quantify a disorder tendency much
better.
R1 R2
R3
R4
R5R5A
R6
R7
R8
R9
R10
R11
R12
R13
R9
P = 3.4
N, H, D = 1.7
N, D = 2.4
N, D, H = 2.0
H, Y, R = 1.6
S, N, D = 1.9
A, E = 1.3
T, H = 1.4
N, D = 2.2
N, H, D = 2.2
N, C, H, D = 2.0
S, A, H = 1.8
S, T = 2.2
V, I, F, Y = 1.3
−180 −120 −90 −60 −30 0 30 60 90 120 150 180
−180 −120 −60 −30 0 30 60 90 120 150 180
R5A
R5
Figure 3: Diagram showing the 14 regions dividing the RP
as proposed by (Kalmankar et al., 2014). Also some amino
acid propensity intensities for inhabiting the RP regions are
depicted.
Using groups of four amino acids taken from cu-
rated proteins from PDB, (Hollingsworth et al., 2012)
explored the existence of recurrent transition patterns
in the RPs. They found 101 significant transitions
between close φ and ψ angles, that represent nearby
configurations visited for many groups of consecu-
tive amino acids. They called these sets (φ, ψ)
2
Mo-
tifs. Figure 4 shows the relevant transitions found by
(Hollingsworth et al., 2012). Although the massive
regions contain the majority of jumps, some of the
motifs are also near to poorly inhabited regions. Un-
fortunately the authors reported that the link between
these motifs and the amino acid sequences, was not
strong enough for identifying direct mapping rules,
making difficult to use the (φ,ψ)
2
Motifs, as a char-
Protein Disorder Prediction using Jumping Motifs from Torsion Angles Dynamics in Ramachandran Plots
41
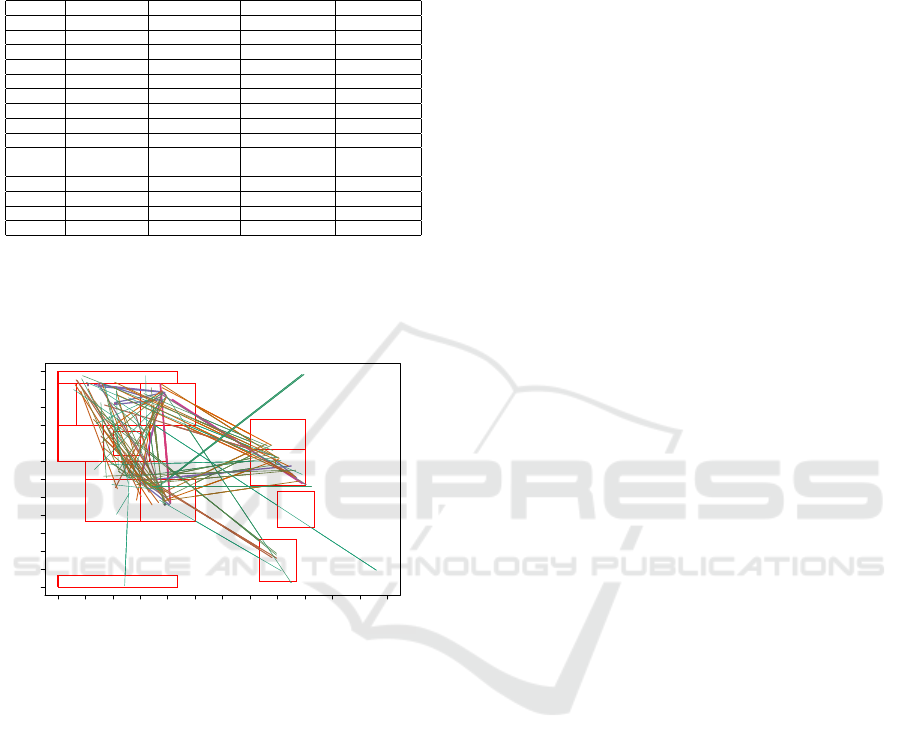
Table 1: Amino acid propensities for inhabiting the 14
zones of the RP as proposed by Kalmankar. Table was
adapted from (Kalmankar et al., 2014). The φ and ψ co-
ordinates ranges are delimiting the zones. Columns 4 and
5 list the high and low propensities of the amino acids for
inhabiting every region.
Region φ ψ High Prop. Low Prop.
1 -160 to -90 90 to 160 V,I,F,Y = 1.3 P = 0.02
2 -90 to -30 90 to 160 P = 3.4
3 -180 to -160 90 to 160 S,A,H =1.8 P,V,L,I = 0.4
4 -180 to -130 30 to 90 N,C,H,D = 2.0 P,I,V,L = 0.4
5 -130 to -80 30 to 90 N,H,D = 1.7 I,V, = 0.5
5A -120 to -90 40 to 80 N,H,D = 2.2 P,V,T = 0.4
6 -150 to -60 0 to 30 N,D = 2.2 P,I,V = 0.5
7 -150 to -90 -70 to 0 T,H = 1.4 P,A = 0.5
8 -90 to -30 -70 to 0 A,E = 1.3
9 -180 to -50 -180 to -160 y S,T = 2.2 L,I = 0.5
160 to 180
10 30 to 90 50 to 100 N,D = 2.4 I,V,L,T = 0.3
11 30 to 90 -10 to 50 N,D,H = 2.0 I,V,T = 0.2
12 60 to 100 -80 to -20 H,Y,R = 1.6 A,L,C = 0.6
13 40 to 80 -170 to -100 S,N,D = 1.9 I,V,T = 0.1
acterization tool for proteins. Therefore, a strategy for
transforming direct amino acid sequences to jumps on
the RP plane, is still missing.
−180 −150 −120 −90 −60 −30 0 30 60 90 120 150 180
−180 −120 −90 −60 −30 0 30 60 90 120 150 180
Figure 4: Graphical representation of the 101 motifs
(φ,ψ)
2
, over the 14 analyzed regions of the RPs. The width
and color of every line, represents the intensity of every mo-
tif. For those motifs with their coordinates lying outside of
the considered regions, the closest zone was selected. Al-
though not all regions are inhabited with the same density,
all of them have been assigned at least one motif.
The amino acid propensities found by (Kalmankar
et al., 2014) and the trajectories implicit in the (φ,ψ)
2
Motifs, can be combined for creating a new protein
characterization, capable of representing indirectly,
the structural transition propensities.
The procedure combines the information present
in the amino acid tendencies for region occupancy
(Table 1), with the initial and final coordinates of the
(φ,ψ)
2
Motifs. Concretely the proteins are taken in
overlapping subsequences of 3 to 5 amino acids. Ev-
ery subsequence has a propensity for inhabit differ-
ent RP regions and these zones are then “activated”.
Then, every activated region, could be the initial or
final target of different (φ,ψ)
2
Motifs. In this way
every subsequence is represented by the intensities of
the activated (φ, ψ)
2
Motifs, times the propensity of
the subchain for inhabiting the activated region. By
this simple procedure, every protein can be mapped to
202 (101 initial plus 101 final) sparse characteristics,
each of them associated to a corresponding (φ,ψ)
2
Motif. We called this strategy, the Jumping Motifs
(jMotifs). The jMotifs characterization is a sparse
representation of every protein sequence, that is in-
directly capturing the dynamic torsion angle propen-
sities along the chain. As an example, Figure 5 is the
representation of a protein, using the jumping Motifs.
In the figure the real state of disorder is signaled
with the black line, when the line is in a high level the
corresponding amino acids are disordered. Is possi-
ble to observe that the disordered regions in this pro-
tein have distinctive activation patterns on the jMotifs
profile. Concretely the first and second disordered re-
gions have a high intensity variation for many jMotifs
when compared with the ordered zones. The pattern
transition is also discernible on the third peak where
the disorder ed zone induces a sustained variation on
the jMotifs intensities. Although these patterns are
not visual identifiable for all the disordered regions in
all the proteins, it will be shown that the jMotifs char-
acterization capture statistically, the transition prefer-
ences in the sequences, allowing the identification of
IDPRs.
2.2 Classification Methods
Structural learning methods are able to model differ-
ent statistical dependences among elements on a se-
quence. This is the case of the probabilistic models
known as Conditional Random Fields (CRFs), which
are able to segment and label sequence data (Lafferty
et al., 2001). The CRFs have several advantages in
comparison to more classical models for sequence
classification such as hidden Markov models. CRFs
belong to the class of discriminative models, so they
model directly the conditional distribution of the la-
bels given the input variables, which is more suitable
for classification purposes. Eq. 1 shows the condi-
tional probability of any particular label y given an
example x used by CRFs. The component F
j
(x,y)
is called a feature function. Intuitively, each feature
function is a specific measure of the compatibility of
the observation x and the label y. Every F
j
(·,·) func-
tion measures a different type of compatibility. The
weighting parameter w
j
, quantifies the influence of
its corresponding feature function in relation with the
other ones. When w
j
> 0, a positive value for the
feature function makes y more likely as the true label
of x. When w
j
< 0, a positive values of the function
BIOINFORMATICS 2018 - 9th International Conference on Bioinformatics Models, Methods and Algorithms
42

1 11 21 31 41 51 61 71 81 91 101 111 121 131 141 151 161 171
MARMNRP AP VE VS YK HMR F L I T HNP TNAT LS T F I EDL K KYGA T T VVRVCE VT YD KT P L E KDG I T V VDWP FDDGAP PPGK VV EDWLS L V KA K FCE APGSCVA VHCV AGL GRAPVL VA L A L I ESGMKYED A I
Q
F I R
Q
KRRGA I NSK
Q
L T Y L EK YRP K
Q
RL R FK DPH THK T RCCVM
1 11 21 31 41 51 61 71 81 91 101 111 121 131 141 151 161 171
MARMNRP AP VE VS YK HMR F L I T HNP TNAT LS T F I EDL K KYGA T T VVRVCE VT YD KT P L E KDG I T V VDWP FDDGAP PPGK VV EDWLS L V KA K FCE APGSCVA VHCV AGL GRAPVL VA L A L I ESGMKYED A I
Q
F I R
Q
KRRGA I NSK
Q
L T Y L EK YRP K
Q
RL R FK DPH THK T RCCVM
Motifs Finales
Final Motifs
Initial Motifs
Figure 5: Jumping Motifs Profile representing protein DP0054 of Disprot. At the top is the activation pattern of initial jMotifs
and below the corresponding activation of final jMotifs. Every jMotif value is along the vertical axis while the amino acid
sequence is along the horizontal axis. Activation intensity is high on the yellow zones and low on the red spots. For this
protein its disorder state is known and is signaled with the black line.
makes y less likely as true label for x. If w
j
= 0 then
the feature function is irrelevant as a predictor of y.
The feature functions are defined in advance by the
designer, while weights are learned by the training al-
gorithm. The denominator Z(x,w) in Eq. 1 is a nor-
malizing factor that constraints the values to the range
[0,1].
P(y|x;w) =
exp
∑
J
j=1
w
j
F
j
(x,y)
Z(x, w)
(1)
In the case of a linear chain CRF, the feature func-
tions must link maximum two labels, while x could
represent any value in the sequence. The Figure 6 de-
picts graphically a linear chain CRF. The convex loss
function used for finding the parameters w
j
, is showed
in Eq. 2. It corresponds to the logarithm of the condi-
tional likelihood associated with Eq. 1. Some advan-
tages of the linear chain CRF, is that convergence to
the global optimum is guaranteed, and efficient train-
ing algorithms do already exist (Lafferty et al., 2001).
LCL(x,y;w) = F
j
(x,y) −
∑
y
0
F
j
(x,y
0
)p(y
0
|x;w) (2)
2.3 Feature Selection
Feature selection can be done independently of the
classification method or can be adjusted to the par-
Figure 6: The graphical structure of the linear chain CRF.
The variable y
t
represents the label in every sequence time-
step. The entire sequence characteristics (x
1
,x
2
,...,x
n
) are
represented in a single node X.
ticular classifier. In this case, we use the regular-
ization parameter of the CRF objective function for
finding a representative subset of characteristics. The
regularization is a strategy for improving model per-
formance, where the objective function that is maxi-
mized during the training phase, is modified for find-
ing fewer or smaller parameters avoiding over-fitting.
It works through the addition of a penalty term, which
castigates the selection of big or abundant parameters.
In the case of CRF, parameters are usually found
by maximizing the log-likelihood function. For exam-
ple, by adding a penalty term based on the L2 norm,
the objective function takes the form showed in Eq. 3.
max
w
LCL(x,y;w) −λw
T
w (3)
max
w
LCL(x,y;w) −λ
∑
i
|
w
i
|
(4)
Protein Disorder Prediction using Jumping Motifs from Torsion Angles Dynamics in Ramachandran Plots
43

In Eq. 3, the parameter λ controls the degree of
the penalty over the likelihood function: parameters
with high magnitude will lead to a higher L2 norm,
reducing the objective function value. Use of the L2-
norm keeps the objective function convex and differ-
entiable, and hence the effort to train a CRF with or
without L2 regularization, is in computationally cost,
very similar. In general, using L2 regularization, the
training procedure will find parameters with reduced
magnitude compared with the function without the
regularization.
A complimentary regularization technique uses
the L1-norm over the parameters. In Eq. 4 the L1
penalty applied over the CRF likelihood function is
shown. In this case, the final model will have many
parameters with exactly zero value, producing simple
and sparse models. L1 regularization applies penal-
ties proportional to parameters magnitude, and al-
though objective function in CRF remains convex, it
is no longer differentiable. This complicates some-
how the training phase when gradient methods are
used (Tsuruoka et al., 2009). The procedure for fea-
ture selection using L1 regularization on CRFs mod-
els is similar to the well known LASSO regression
(Tibshirani, 1996). A regularization path is recon-
structed using different values of λ in the regular-
ization term. The regularization path allows the in-
formed selection of a given set of characteristics, ac-
cording to the performance reached. For avoiding
over fitting and finding the appropriate characteristics,
the regularization path must be found on different par-
titions of the data. That is, a hold out set is required
for allowing correct validation of the characteristics
found. Concretely the algorithm used for finding the
selected features was:
Feature Selection on CRF using L1-norm:
1. Partition of dataset in Train, Test and Valida-
tion subsets
2. Find λ
0
, the value of L1 regularization pa-
rameter that excludes all the properties
3. While some weight = 0
3.1. Decrease λ (this allows to include proper-
ties progressively)
3.2. Train CRF using the Train set and apply-
ing L1 regularization with λ parameter
3.3. Test the CRF using the test set
3.4. Compute performance metrics for Test
and Train samples
3.5. End while
4. Find best λ considering metrics on Test sam-
ples
5. Select only the features present when using
best Lambda on test samples
6. Train CRF model using Train + Test samples
with the features selected, and L2 Regular-
ization
7. Test CRF using Validation samples if avail-
able
2.3.1 Properties Selected
The procedure using the regularization path for select-
ing the best model characteristics, was implemented
on the 202 jMotifs. Initially, using a shallow training
(12 iterations), the value of parameter λ that excluded
all the properties was explored. This was called λ
0
,
and found to be the value 10
5.3
. Later, exploring
λ from 10
−1
to λ
0
, regularizations paths were com-
puted. Figure 7 shows the AUC performance, the
magnitude of weights and the number of variables in-
cluded as λ parameter varies in a logarithm progres-
sion. The point where AUC performance in training
samples reaches a plateau, is selected as the optimal
point for variable inclusion. Thus, 37 of the original
202 properties were included.
−1 0 1 2 3 4 5
0.4
0.45
0.5
0.55
0.6
0.65
0.7
0.75
0.8
0.85
0.9
X: 2.4
Y: 0.8382
Testing
Training
−1 0 1 2 3 4 5
0
2
4
6
8
10
Log10(Lambda)
−1 0 1 2 3 4 5
0
100
200
300
X: 2.4
Y: 37
Número de variables activas al Variar Factor Regularización L1
Log10(Lambda)
AUC
Log(
)
Log(
)
Log(
)
Weights sum of variables included
Number of variables included
Figure 7: Regularization path with jMotif properties. AUC
metric on training and testing samples is on the left. In the
point where log(λ) = 2.4, are required only 37 character-
istics, as can be see in the right plot, with these properties
subset is enough for reaching a good performance.
Positions of the selected jMotifs appear on Fig-
ure 8. We can see that many of the 14 RPs zones are
represented for the selected jMotifs. There are four
jMotifs that conserved its initial and final points, these
were ββ.1, PP.1, Pδ.1 and Pδ.2. Transitions in region
β and P, have the same zone as origin and destination,
it means they represent amino acids that preserve their
torsional configurations in the jump. In Figure 8 the
transitions given by jumps from P to δ
0
and δ
0
to P
0
,
are represented using arrows. It is quite notorious that
symmetrical jumps, are marked as important for the
identification of disorder. Additionally P
0
is a low in-
habited region, and nevertheless it is identified as rel-
evant for the recognition of disordered amino acids.
BIOINFORMATICS 2018 - 9th International Conference on Bioinformatics Models, Methods and Algorithms
44

φ
ψ
φ
a)
b)
Figure 8: a) RP coordinates of initial (blue) and final (black) selected jMotifs with asymmetrical components. Important areas
of the RPs are covered by the selected jMotifs, even some low inhabited regions are represented. b) For some jMotifs, both
components were selected, and final and initial coordinates are shown. Two of these transitions involves jumps in the same
area.
3 EXPERIMENTS AND RESULTS
For the sake of comparison, the proposed charac-
terization methods were evaluated on a target data
set and their result were compared with sequence
based predictors and also with MSA based methods.
The CRF models were implemented using the library
HCRF2.0b ((Morency, 2015)).
3.1 Data Sets
For training our predictor, we used the 3000 se-
quences in the database DM3000, prepared in (Zhang
et al., 2012). This dataset mainly contains proteins
took from PDB and selected with the following cri-
teria: a resolution less than 2 amstrongs, a size big-
ger than 60 residues and having X-ray structures with
missing electron densities for groups of amino acids,
which are assumed to be in disorder.
Later, the proposed predictor was evaluated on the
SL329 Data set, which was prepared in (Zhang et al.,
2012). The referenced authors created the database
selecting proteins with sequence homology less than
(25%) from the SL benchmark data set. The SL data
set (Sirota et al., 2010) is a subset of Disprot, the
most referenced and commonly used database (Sick-
meier et al., 2007). SL329 contains 329 proteins
with 51.292 ordered residues and 39.544 disordered
residues.
3.2 Model Validation
The selection of model parameters was carried out
using a 10-fold cross-validation strategy on train-
ing samples. In general data sets can include
some level of imbalance between ordered and dis-
ordered proteins, then some metrics able to quan-
tify the performance in such scenarios were included.
The set of metrics used includes: AUC, Sensitiv-
ity, Specificity, B
ACC
and MCC. AUC refers to the
area under the ROC curve, being disorder the pos-
itive class. MCC is the Matthews correlation co-
efficient, which according to (Baldi et al., 2000) is
an appropriate measure of performance for unbal-
anced data sets. MCC can be estimated as MCC =
T P·TN−FP·FN
√
(T P+FP)·(T P+FN)·(T N+FP)·(T N+F N)
, where TP de-
notes True Positive, TN stands for True Negative, FP
is False Positive and FN is False Negative.
On the other hand, B
ACC
is the balanced accuracy
which can be expressed as
B
ACC
=
Sensi + Speci
2
(5)
where Sensi = T P/(T P + FN), and Speci =
T N/(T N + FP) are the sensitivity and the specificity
respectively.
3.3 Results
Table 2 shows evaluation results on benchmark SL329
data set. Performance of compared methods were
took from (Faraggi et al., 2012). Is possible to observe
Protein Disorder Prediction using Jumping Motifs from Torsion Angles Dynamics in Ramachandran Plots
45

that the method SPINE-D, who is using sequence
alignment, obtained the best performance on this
data set. The proposed jMotCRiF method achieved
the second best performance, surpassing many MSA
methods in the state-of-art. Even metapredictors as
MFdp and MD, are doing comparatively worse on this
data set than jMotCRiF. Considering that jMotCRiF
is using only information from the RPs, this per-
formance is quite impressive. We can also observe
that all the free alignment methods were surpassed.
For example jMotCRif gets an AUC 4.5% bigger
than commonly referenced IUPRED. Espritz showed
a good performance compared with many MSA-
based methods, but its metrics did not surpassed the
metapredictors or jMotCRiF. In terms of MCC and
AUC, jMotCRiF outperforms Espritz in about 2.5%
and 1.6% respectively, considering relative differ-
ences. jMotCRiF outperforms some of the state-of-
art MSA-based methods, with a considerably margin,
for example MCC metric of jMotCRiF is 63% higher
that the same value in PONDR.
The performance of SPINE-D is better, although
pretty close to the one obtained by jMotCRiF. This
result could be explained due to the fact that SPINE-
D corresponds to an adaptation of a secondary struc-
ture predictor, which was based on the prediction of
torsion angles from sequence profiles (Faraggi et al.,
2012). jMotCRiF is also using information of torsion
angles, but applying a more simple strategy which
is based only in the protein sequence, without us-
ing MSA algorithms and the collection of proper-
ties that SPINE-D requires, as the prediction of sec-
ondary structure, complexity, amino acid composi-
tion, physic-chemical characteristics, etc.
Table 2: Performance comparison among disorder identifi-
cation methods on SL329 data set. jMotCRiF reaches the
second place in AUC value, even without using MSA algo-
rithms.
METHOD AUC SEN SPE MCC TYPE
SPINE-D 0,886 0,780 0,850 0,630 MSA
jMotCRiF 0,877 0,804 0,824 0,621 FREE
MFDp 0,873 0,880 0,620 0,510 MSA
MD 0,864 0,660 0,890 0,580 MSA
Espritz 0,863 0,728 0,868 0,606 FREE
Disopred 0,858 0,690 0,900 0,590 MSA
PONDR 0,843 0,610 0,910 0,550 MSA
IUPRED 0,839 0,758 0,598 0,504 FREE
NORSnet 0,815 0,540 0,920 0,510 MSA
PONDR 0,755 0,590 0,780 0,380 MSA
4 DISCUSSION AND
CONCLUSIONS
In this paper, a new methodology for characterizing
protein sequences that rely exclusively in the occu-
pation propensities on the Ramachandran plots was
described. The strategy aims to capture, at least in-
directly, the dynamic variations of the torsional an-
gles in the amino acid chains, for creating suitable nu-
merical descriptors that can be linked with the amino
acid disorder state. Using this fast characterization, a
classification based on structured classifiers was ex-
plored and tuned. The obtained predictor, jMotCRiF,
is a fast and alignment-free tool for disorder identifi-
cation, that is capable of achieving high performance
when compared with the state-of-art methods.
Even though jMotCRiF could be use as stand
alone predictor, the results obtained show that there
is still an improvement margin to be reached. An al-
ternative for attaining a better performance, could be
the combination of jMotCRiF with other complemen-
tary features available in the state of the art, or with
current high potential classification techniques such
as different deep learning architectures. In the past,
we proposed a predictor, CRF InfoThor (Uribe et al.,
2017), also inspired in the dynamics hidden on the
RPs, which also reached a good performance in the
identification of disordered regions, although it was
based on more complex descriptors. Either in combi-
nation with that predictor or with some of the classi-
fiers in the state-of-art, jMotCRiF has the potential for
obtaining a high performance and contribute with the
correct labeling of IDPRs. Additional experiments
for validating such progress must be done on bigger
datasets and with the inclusion of the different disor-
der predictors for achieving an appropriate compari-
son.
REFERENCES
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and
Lipman, D. J. (1990). Basic local alignment search
tool. Journal of molecular biology, 215(3):403–410.
Altschul, S. F., Madden, T. L., Schffer, A. A., Zhang, J.,
Zhang, Z., Miller, W., and Lipman, D. J. (1997).
Gapped BLAST and PSI-BLAST: a new generation
of protein database search programs. Nucleic acids
research, 25(17):3389–3402.
Baldi, P., Brunak, S., Chauvin, Y., Andersen, C. A., and
Nielsen, H. (2000). Assessing the accuracy of predic-
tion algorithms for classification: an overview. Bioin-
formatics, 16(5):412–424.
Baruah, A., Rani, P., and Biswas, P. (2015). Conforma-
tional entropy of intrinsically disordered proteins from
amino acid triads. Scientific reports, 5.
Berman, H., Henrick, K., Nakamura, H., and Markley, J. L.
(2007). The worldwide Protein Data Bank (wwPDB):
ensuring a single, uniform archive of PDB data. Nu-
cleic acids research, 35(suppl 1):D301–D303.
BIOINFORMATICS 2018 - 9th International Conference on Bioinformatics Models, Methods and Algorithms
46

Bulashevska, A. and Eils, R. (2008). Using Bayesian multi-
nomial classifier to predict whether a given protein se-
quence is intrinsically disordered. Journal of theoret-
ical biology, 254(4):799–803.
Cortes, C. and Vapnik, V. (1995). Support-vector networks.
Machine Learning, 20(3):273–297.
DeForte, S. and Uversky, V. N. (2016). Order, Disorder, and
Everything in Between. Molecules, 21(8):1090.
Deng, X., Eickholt, J., and Cheng, J. (2012). A compre-
hensive overview of computational protein disorder
prediction methods. Molecular BioSystems, 8(1):114–
121.
Dosztnyi, Z., Csizmok, V., Tompa, P., and Simon, I. (2005).
IUPred: web server for the prediction of intrinsically
unstructured regions of proteins based on estimated
energy content. Bioinformatics, 21(16):3433–3434.
Faraggi, E., Zhang, T., Yang, Y., Kurgan, L., and Zhou, Y.
(2012). SPINE X: improving protein secondary struc-
ture prediction by multistep learning coupled with
prediction of solvent accessible surface area and back-
bone torsion angles. Journal of computational chem-
istry, 33(3):259–267.
He, B., Wang, K., Liu, Y., Xue, B., Uversky, V. N., and
Dunker, A. K. (2009). Predicting intrinsic disorder in
proteins: an overview. Cell research, 19(8):929–949.
Hollingsworth, S. A. and Karplus, P. A. (2010). A fresh look
at the Ramachandran plot and the occurrence of stan-
dard structures in proteins. Biomolecular concepts,
1(3-4):271–283.
Hollingsworth, S. A., Lewis, M. C., Berkholz, D. S., Wong,
W.-K., and Karplus, P. A. (2012). ($\phi$, $\psi$)
2 motifs: a purely conformation-based fine-grained
enumeration of protein parts at the two-residue level.
Journal of molecular biology, 416(1):78–93.
Jones, D. T. and Cozzetto, D. (2014). DISOPRED3: precise
disordered region predictions with annotated protein-
binding activity. Bioinformatics, page btu744.
Kalmankar, N. V., Ramakrishnan, C., and Balaram, P.
(2014). Sparsely populated residue conformations
in protein structures: Revisiting experimental Ra-
machandran maps. Proteins: Structure, Function, and
Bioinformatics, 82(7):1101–1112.
Kawashima, S. and Kanehisa, M. (2000). AAindex:
amino acid index database. Nucleic acids research,
28(1):374–374.
Kozlowski, L. P. and Bujnicki, J. M. (2012). MetaDisorder:
a meta-server for the prediction of intrinsic disorder in
proteins. BMC bioinformatics, 13(1):111.
Lafferty, J., McCallum, A., and Pereira, F. (2001). Con-
ditional random fields: Probabilistic models for seg-
menting and labeling sequence data. In Proceedings
of the eighteenth international conference on machine
learning, ICML, volume 1, pages 282–289.
Lieutaud, P., Canard, B., and Longhi, S. (2008). MeDor: a
metaserver for predicting protein disorder. BMC ge-
nomics, 9(Suppl 2):S25.
Mizianty, M. J., Stach, W., Chen, K., Kedarisetti, K. D.,
Disfani, F. M., and Kurgan, L. (2010). Improved
sequence-based prediction of disordered regions with
multilayer fusion of multiple information sources.
Bioinformatics, 26(18):i489–i496.
Morency, L.-P. (2015). HCRF library (including CRF and
LDCRF).
Oates, M. E., Romero, P., Ishida, T., Ghalwash, M.,
Mizianty, M. J., Xue, B., Dosztnyi, Z., Uversky, V. N.,
Obradovic, Z., Kurgan, L., and others (2013). D2p2:
database of disordered protein predictions. Nucleic
acids research, 41(D1):D508–D516.
Peng, Z., Yan, J., Fan, X., Mizianty, M. J., Xue, B., Wang,
K., Hu, G., Uversky, V. N., and Kurgan, L. (2015).
Exceptionally abundant exceptions: comprehensive
characterization of intrinsic disorder in all domains of
life. Cellular and Molecular Life Sciences, 72(1):137–
151.
Potenza, E., Di Domenico, T., Walsh, I., and Tosatto, S. C.
(2015). MobiDB 2.0: an improved database of intrin-
sically disordered and mobile proteins. Nucleic acids
research, 43(D1):D315–D320.
Schlessinger, A., Punta, M., and Rost, B. (2007). Natively
unstructured regions in proteins identified from con-
tact predictions. Bioinformatics, 23(18):2376–2384.
Sickmeier, M., Hamilton, J. A., LeGall, T., Vacic, V.,
Cortese, M. S., Tantos, A., Szabo, B., Tompa, P.,
Chen, J., Uversky, V. N., and others (2007). DisProt:
the database of disordered proteins. Nucleic acids re-
search, 35(suppl 1):D786–D793.
Sirota, F. L., Ooi, H.-S., Gattermayer, T., Schneider, G.,
Eisenhaber, F., and Maurer-Stroh, S. (2010). Parame-
terization of disorder predictors for large-scale appli-
cations requiring high specificity by using an extended
benchmark dataset. BMC genomics, 11(Suppl 1):S15.
Tibshirani, R. (1996). Regression shrinkage and selection
via the lasso. Journal of the Royal Statistical Society.
Series B (Methodological), pages 267–288.
Tsuruoka, Y., Tsujii, J., and Ananiadou, S. (2009). Stochas-
tic Gradient Descent Training for L1-regularized Log-
linear Models with Cumulative Penalty. In Pro-
ceedings of the Joint Conference of the 47th Annual
Meeting of the ACL and the 4th International Joint
Conference on Natural Language Processing of the
AFNLP: Volume 1 - Volume 1, ACL ’09, pages 477–
485, Stroudsburg, PA, USA. Association for Compu-
tational Linguistics.
Uribe, J. A., Arias-Londo
˜
no, J. D., and Perera-Lluna, A.
(2017). Protein Disorder Prediction using Informa-
tion Theory Measures on the Distribution of the Dihe-
dral Torsion Angles from Ramachandran Plots:. pages
43–51. SCITEPRESS - Science and Technology Pub-
lications.
Uversky, V. N., Oldfield, C. J., and Dunker, A. K. (2008).
Intrinsically disordered proteins in human diseases:
introducing the D2 concept. Annu. Rev. Biophys.,
37:215–246.
Varadi, M., Vranken, W., Guharoy, M., and Tompa, P.
(2015). Computational approaches for inferring the
functions of intrinsically disordered proteins. Fron-
tiers in molecular biosciences, 2.
Walsh, I., Martin, A. J., Di Domenico, T., and Tosatto, S. C.
Protein Disorder Prediction using Jumping Motifs from Torsion Angles Dynamics in Ramachandran Plots
47

(2012). ESpritz: accurate and fast prediction of pro-
tein disorder. Bioinformatics, 28(4):503–509.
Wang, L. and Sauer, U. H. (2008). OnD-CRF: predicting or-
der and disorder in proteins conditional random fields.
Bioinformatics, 24(11):1401–1402.
Ward, J. J., Sodhi, J. S., McGuffin, L. J., Buxton, B. F., and
Jones, D. T. (2004). Prediction and functional analysis
of native disorder in proteins from the three kingdoms
of life. Journal of molecular biology, 337(3):635–645.
Xue, B., Dunbrack, R. L., Williams, R. W., Dunker,
A. K., and Uversky, V. N. (2010). PONDR-FIT: a
meta-predictor of intrinsically disordered amino acids.
Biochimica et Biophysica Acta (BBA)-Proteins and
Proteomics, 1804(4):996–1010.
Zhang, T., Faraggi, E., Xue, B., Dunker, A. K., Uversky,
V. N., and Zhou, Y. (2012). SPINE-D: accurate predic-
tion of short and long disordered regions by a single
neural-network based method. Journal of Biomolecu-
lar Structure and Dynamics, 29(4):799–813.
BIOINFORMATICS 2018 - 9th International Conference on Bioinformatics Models, Methods and Algorithms
48
