
An in Silico Approach for Understanding the Complex Intercellular
Interaction Patterns in Cancer Cells
Maura Cárdenas-García
1
and Pedro Pablo González-Pérez
2
1
Facultad de Medicina, Benemérita Universidad Autónoma de Puebla, 13 Sur 2702, Puebla, Mexico
2
Departamento de Matemáticas Aplicadas y Sistemas, Universidad Autónoma Metropolitana,
Unidad Cuajimalpa, Ciudad de México, Mexico
Keywords: In Silico Approach, Modelling and Simulation, Intercellular Interaction, Cancer Cells, Signalling Pathways.
Abstract: Intercellular interaction allows cancer cells to preserve their malignance and through cell junctions to induce
malignance in neighbouring cells and receive nutrients from them. The Wnt (wingless-related integration
site) signalling pathway plays an important role in the formation of intercellular communications. In this
work, we explore the complex interactions patterns of intercellular communication in cancer cells using an
in silico modelling and simulation methodology developed by us. The proposed cellular signalling model,
characterized by a multicompartmental nature, provides symbolic abstractions and accurate algorithms to
model both intracellular and intercellular behaviours. In particular, in this work, we propose an in silico
model and simulation of the formation of different communication channels, involving the Wnt signalling
pathway. The final purpose of this study is to propose target molecules leading to break the communication
between a cancer cell and surrounding normal cells. In this way, it is not necessary to carry out long series
of different in vitro experiments, but only a few, because the focus should be only on the key molecules,
which saves time and money. We observed, using in silico experiments, how the inhibition of Wnt
signalling pathway prevents that the cells surrounding a cancerous cell are transformed.
1 INTRODUCTION
Intercellular communication allows the transfer of
information from one cell to another. This type of
interaction takes place by physical unions between
the cells or through signalling molecules, released in
the extracellular space by one cell and received by
another cell through its receptors. By means of
intercellular communication, the cells work in
coordination, facilitating their survival. In the case
of cancer cells, this communication allows them to
continue growing, as well as inducing malignancy in
neighbouring cells (Brücher and Jamall, 2014).
Surrounding normal cells also send signals to cancer
cells, thus these are destroyed, but not always
successfully. The formation of junctions between
normal and malignant cells plays an important role
in the control of a cellular focus that will result in a
tumour (Lum and Chen, 2015). The Wnt signalling
pathway plays an important role in the formation of
intercellular communications and regulates cell
proliferation. The Wnt protein family includes a
large number of cysteine-rich glycoproteins. Wnt
proteins activate different signal transduction
pathways. In this work, we model and simulate the
canonical Wnt signalling pathway because this
pathway is related to other signalling pathways in
cancer.
The modelling and simulation of cellular
signalling systems has found valuable support in a
wide range of modelling approaches, which cover a
wide spectrum ranging from mathematical models,
e.g., ordinary differential equation systems,
statistical methods and numerical methods, to
computational models, e.g., cellular automata,
Boolean networks, Petri nets, neural networks and
multi-agent systems. Based on these models,
different simulation tools have been developed from
mathematical ones to computational ones (Alves et
al., 2006). However, the majority of these former
tools are based on abstractions to model only
intracellular behaviour. Thus, they are not suitable to
model intercellular communication. Consequently,
in recent years, other major requirements in the
simulation of cellular communication systems –such
as multiple levels of compartmentalization, topology
188
Cárdenas-García, M. and Pérez, P.
An in Silico Approach for Understanding the Complex Intercellular Interaction Patterns in Cancer Cells.
DOI: 10.5220/0006722601880195
In Proceedings of the 11th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2018) - Volume 3: BIOINFORMATICS, pages 188-195
ISBN: 978-989-758-280-6
Copyright © 2018 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

and locality– have emerged, guiding the
development of new computational models and
tools. Notable examples of computational simulation
tools supporting such features are Bio-PEPA
(Ciocchetta et al., 2009), MCell (Kerr et al., 2008),
COPASI (Hoops et al., 2006), Virtual Cell (Cowan,
2012) and CompuCell 3D (Swat et al., 2012).
In this work, we simulate and explore the
complex interaction patterns of intercellular
communication in cancer cells using an in silico
modelling and simulation approach developed by us,
and motivated by two key elements: 1) a tuple
space-based model (Gelernter, 1985) for the
representation of the signalling elements (i.e.,
reactions with their own kinetic parameters, and
reactants with their concentrations) and 2) a control
mechanism for the selection and execution of
reactions based on the Gillespie’s algorithm
(Gillespie, 1971) –a stochastic simulation algorithm
typically used to mimic systems of
chemical/biochemical reactions in an efficient and
accurate way. A significant characteristic of this in
silico approach is its multicompartmental nature.
Specifically, it is suitable to model cells in their
“social context”, along with all those biological
mechanisms that involve two or more cells, that is
essential in the scenario discussed in this work. The
main idea behind of this in silico modelling and
simulation approach is to provide a computational
experimentation environment that complements and
guides in vitro experimentation in intra and
intercellular signalling networks.
In this work, we showed and discussed how this
in silico modelling and simulation approach can be
used successfully to understand and explore the
intercellular communication between a cancer cell
and normal cells and, consequently, to propose key
molecules, which can be targeted to allow us to
break the communication between cancer cells and
surrounding normal cells.
2 MATERIALS AND METHODS
2.1 The Multicompartmental
Intercellular Signalling Model
Here we introduce the key elements of the intra and
intercellular signalling model, characterized by its
multicompartmental nature. The proposed model
provides the logical abstractions for the
representation and manipulation of the signalling
elements, i.e., reactions, reactants and products. As
mentioned earlier, the logical abstractions for the
representation of the signalling elements are
conceived from the notion of tuple and tuple spaces
(Gelernter, 1985; González-Pérez et al, 2013),
although the control mechanism for the selection and
execution of the reactions is based in Gillespie's
algorithm.
2.1.1 Tuple Space-based Model for
Representation of Signalling Elements
Denote by C
i
, 1 ≤ i ≤ m, the i-th cell belonging to the
cellular group G, which is represented by a set of n
tuple spaces (TS) such that:
(1)
Each tuple space TS
ij
, 1 ≤ j ≤n, is a set of tuples,
where each individual tuple (t) represents a
signalling element. Denote by cr a reaction, by r a
reactant, and by p a product, therefore we have:
(2)
From (1) and (2) we have that any tuple t in any
tuple space TS
ij
, 1 ≤ j ≤n, and therefore in cell C
i
,
represents either a reaction (cr), a reactant (r) or a
product (p). Note that each TS
ij
, 1 ≤ j ≤n, represents
a cell compartment, e.g., nucleus, mitochondria,
cytoplasm, cell membrane, or even extracellular
space, which guarantees the multicompartmental
nature of the cell signalling model.
In order to simplify the notation, the subscripts
corresponding to cr, r and p have not been
considered in expression (2). However, it should be
clear that each of these identifiers is accompanied by
three sub-indices, the first refers to the set of spaces
of tuples (cell), the second refers to the space of
tuples (cellular compartment), and the third refers to
the element itself (particular reaction, reactant o
product).
Regarding representation of reactions,
expression (3) provides the symbolic abstraction that
allows to represent, and therefore manipulate,
chemical reaction schemes commonly required when
modelling cellular signalling systems, such as
synthesis, decomposition, and standard equation for
enzymatic reactions, as referred in expression (4) to
(6), respectively.
(3)
where: r
1
, r
2
are reactants and reqMol
1
, reqMol
2
are
the number of molecules involved of reactants r
1
, r
2
,
respectively; K is the reaction rate constant; p
1
, p
2
are products and pm
1
, pm
2
are the number of
molecules formed of products p
1
, p
2
, respectively.
An in Silico Approach for Understanding the Complex Intercellular Interaction Patterns in Cancer Cells
189

(4)
(5)
(6)
Let TS
ij
and TS
ik
be two neighbouring tuple
spaces, which we represent by the tuple:
(7)
Consider also that a tuple space can have at most
two neighbours, given the type of biological system
that we are modelling. As already established, a
tuple space (TS
ij
) models a particular cellular
compartment. Thus, an example of tuple space with
more than one neighbour is given by the “cellular
membrane” tuple space, which has as neighbours the
“extracellular space” and the “cytosol” tuple spaces.
The notion of neighbouring tuple spaces
(expression (7)) plays a key role in our signalling
model, since it allows us to establish that the
products formed by a reaction cr belonging to a
tuple space TS
ij
, are translocated to another tuple
space TS
ik
, if and only if TS
ij
and TS
ik
are neighbours.
In this way, the continuity of the signal transduction
is guaranteed through all tuple spaces (cell
compartments) that make up the cell C
i
.
Then, returning to expression (3), if we require
that one of the products formed, for example p
1
, be
translocated to the tuple space TS
ik
, being TS
ij
and
TS
ik
neighbours, then the tuple (p
1
, pm
1
), located in
the right part of the expression (3) will be replaced
by the tuple (translocate(p
1
, pm
1
), TS
ik
).
With regard to reactants and products involved
in the reactions, both are also represented as tuples
in the tuple space, through the symbolic abstraction
provided in the expression (8).
(8)
where r
i
is the reactant and Mol
i
is the number of
available molecules.
2.1.2 The Algorithm for the Selection and
Execution of Reactions
Once all the reactions and the reactants are
modelled, then every reaction is explicitly simulated
on the basis of the Gillespie algorithm. In detail, the
main steps performed by the algorithm for the
selection and execution of reactions, i.e., for starting
and continuing cellular signal transduction, are
summarized below:
1. Calculate the rate for each eligible reaction
(cr) –see expression (3)– according to the
expression:
(9)
where K is the reaction rate constant, Mol
i
is the
number of available molecules of reactant r
i
, and
reqMol
i
is the number of molecules required of
reactant r
i.
The rate with which the reaction will be selected
is equal to the rate of this reaction (K) –where K can
be estimated as a measure of affinity or calculated
from the maximum rate (V
max
) and the Michaelis
constant (K
m
)– multiplied by the product of the
binomial coefficients of the available moles of each
reactant involved in the reaction and the number of
moles of this required by the reaction. If a reaction is
not eligible for lack of any of the required reactants,
then the rate (Rate) of this reactions will be zero.
2. Calculate the summation of the rates (Rate) of
all eligible reactions, the resulting value is RTot.
3. Sort all eligible reactions by rate in a
descending order.
4. Generate a random number ψ between 0 and 1.
5. From sorted list of eligible reactions, the k-th
reaction is chosen if:
(10)
Note, that the value of the summation is equal to 1
for the last reaction in the sorted list. So, if there are
eligible reactions, then one of them will always be
executed.
6. Generate a random number τ between 0 and 1.
Stop the execution of the reactions for a time
given by:
(11)
The simulation proceeds choosing the next
reaction to occur on the basis of a random number
and its propensity function that is calculated based
on the reaction rate and on the number of reactants.
The time interval to update the simulation time is
also computed step by step depending on a random
number and on the sum of propensity functions of all
reactions. The iteration of these steps (involving
expressions (9), (10) and (11)) constitutes the
simulation. The simulation concludes when there are
no eligible reactions.
BIOINFORMATICS 2018 - 9th International Conference on Bioinformatics Models, Methods and Algorithms
190
2.2 The Computational Simulation
Tool Associated with the Proposed
Model
The modelling approach proposed was integrated in
Cellulat, an already existing computational
simulation tool for signal transduction systems,
developed by us (González-Pérez et al., 2013;
Cárdenas-García et al., 2016). Cellulat establishes in
itself an integrated virtual environment for in silico
experimentation on cellular signalling pathways and
networks, strongly dependent on characteristics such
as multi-compartmentalization, location and
topology.
As a highly interactive application, Cellulat
provides the user with a wide range of visual and
interactive tools, to follow and feedback at every
moment the signal transduction that takes place in
the simulated signalling network.
It is important to note that all the elements
required by the simulation –cellular structures and
compartments, reactions with their kinetic
parameters, and reactants with their concentration or
number of available molecules– are written or
recorded in the same written language used when
describing these elements; it is the simulation tool
itself that will translate them into logical abstractions
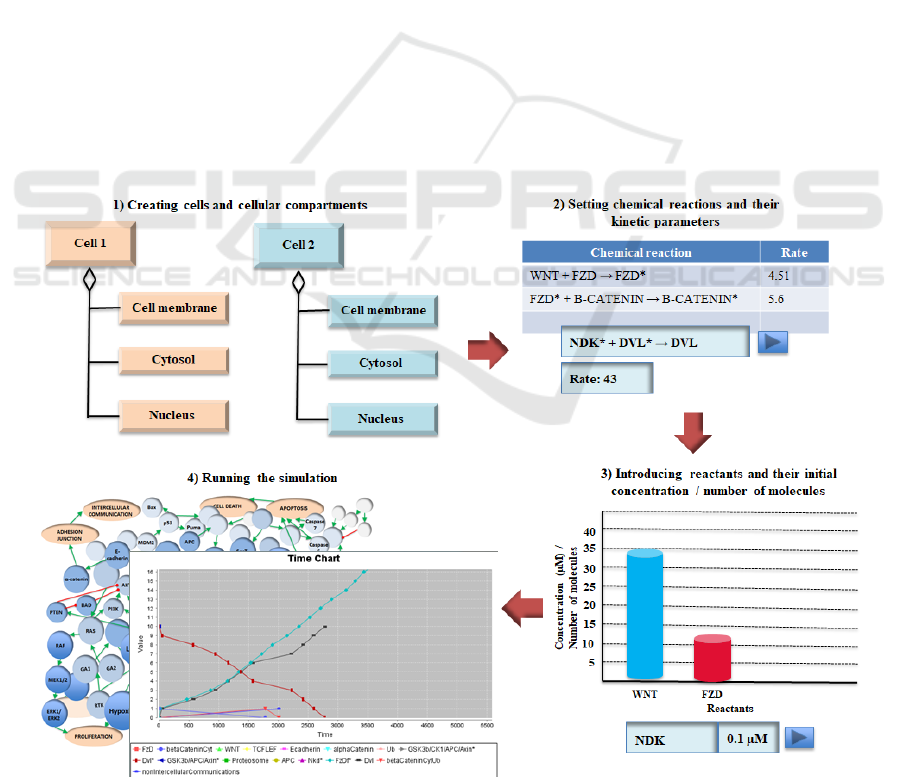
based on tuple spaces, previously introduced. Figure
1 shows the key phases in the creation and execution
of a simulation using Cellulat.
Cellulat (in its Executable Jar File version) can
be either executed or downloaded from the
bioinformatics website of our research group at
http://bioinformatics.cua.uam.mx/node/10. The
instructions required for the download can be
consulted on this website.
2.3 The Methodological Approach
The methodology followed in this work is based on
a continuous bidirectional feedback between the in
silico modelling and simulation approach and
theoretical and experimental knowledge. That is, the
proposed multicompartmental intercellular signal-
ling model and the results of its associated
computational simulation should provide valuable
support to guide in vitro experimentation; while the
results of theoretical and experimental research
should lead to both the improvement of the model
and the design of the most appropriate in silico
experiments.
Figure 1: Sequence of the major activities carried out during the creation and execution of a cell signalling simulation using
Cellulat.
An in Silico Approach for Understanding the Complex Intercellular Interaction Patterns in Cancer Cells
191

The main phases involved in this methodology are
summarized below:
Modelling phases:
1. Modelling the integrated Wnt signalling
pathways.
2. Taking into account the model developed in
phase 1, define the cellular structures and
compartments involved in the signalling and,
for each of these, the list of reactions and
reactants located there.
Simulation phases:
3. Creating cellular compartments.
4. Setting reactions and their kinetic parameters.
5. Introducing reactants and their available
concentration o number of molecules.
6. Design the in silico experiments.
7. Running the simulation.
8. Visualize simulation execution.
Analysis phases:
9. Analysis of simulation execution.
10. Feedback between the in silico modelling and
simulation approach and theoretical and
experimental knowledge.
3 RESULTS
3.1 The Integrated Model of Wnt
Signalling Pathways
Figure 2 shows, as first result, a simplified version
of Wnt signalling pathway model proposed by us, to
be simulated using the in silico approach previously
presented and following the methodology described
above. As can be seen in Figure 2, β-catenin is the
central part in this pathway, interacting with E-
cadherin and α-catenin proteins. We have integrated
into the model the canonical and non-canonical
pathways as well as Notch (Borggrefe et al., 2016),
Hedgehog (Shimizu and Nakagawa, 2015), and
Hypoxias (Grunsven and Vlierberghe, 2014) ones.
On the other hand, we will also consider, as the
background of the model illustrated in Figure 2,
other previously modelled signalling pathways with
which the Wnt signalling path intersects, such as
EGF/MAPK/JAK-STAT (González-Pérez et al.,
2003), PI3K (González-Pérez et al., 2013) and
caspases (Cárdenas-García and González-Pérez,
2013). In Table 1 can be observed some examples of
signalling elements –reactions and reactants–
defined as part of the integrated model of Wnt
signalling pathways. Note that both types of cells
(normal and cancer cells) exhibit the same signalling
elements to form communication channels.
Figure 2: A simplified version of Wnt signalling pathway
modelled in this study. Red arrows indicate inhibition
relationships and green arrows indicate activation
relationships.
3.2 The Simulation of the Wnt
Signalling Pathways
As can be seen in Figure 3, the simulation is ready
for execution. That is, cellular compartments,
reactions and reactants have been created from the
Wnt signalling pathway model, which has been
conceived and verified as initial phase of our
methodological approach.
The simulated Wnt signalling network is made
up of 146 nodes representing reactants, 12 nodes
representing cell processes, such as adhesion
junction, proliferation, apoptosis and cell death, and
204 arcs representing reactions between the involved
nodes. Table 1 shows some reaction examples. The
overall signalling network extends across 5 cell
compartments (i.e., extracellular space, cell
membrane, cytosol, mitochondria and nucleus)
comprising key cellular signalling pathways
involved in growth and metabolism leading to
survival, proliferation, tumour progression and cell
death, as well as integration with the formation of
intercellular interactions (i.e., EGF/MAPK/JAK-
STAT, PI3K/AKT and caspases signalling
pathways).
BIOINFORMATICS 2018 - 9th International Conference on Bioinformatics Models, Methods and Algorithms
192

Table 1: Examples of signalling elements –reactions and reactants– defined as part of the integrated model of Wnt
signalling pathways.
Cellular
compartment
Reaction
Vmax
Reactant
Reactant
Conc. (µM)
Reference
Extracellular
space / membrane
WNT + FZD -> FZD
*
4.51
FZD
WNT
12
33
(Lee et al., 2003)
Cytosol
FZD
*
+ β-CATENIN ->
β-CATENIN
*
5.6
β-CATENIN
0.001
(Hernández et al.,
2012)
Cytosol
NDK
*
+ DVL
*
-> DVL
43
NDK
DVL
0.1
0.01
(Blaheta et al. 2005)
Cytosol
APC
*
+ β-CATENIN
*
->
β-CATENIN
12
APC
0.4
(Blaheta et al. 2005)
Cytosol
β-CATENIN
*
+ TCF-LEF ->
β-CATENIN
*
/ TCF-LEF
33
TCF-LEF
8
(Hernández et al.,
2012)
Figure 3: Cells, cell compartments, reactions and reactants have been created as the initial components required by the
simulation of Wnt signalling pathways. Signalling elements, e.g., proteins and enzymes, are represented by solid blue
spheres. Each signalling element is identified by its name (acronym). Red arrows indicate inhibition relationships and green
arrows indicate activation relationships.
3.3 An Overview of the in Silico
Experiments
The simulated intercellular communication scenario
consisted of two cells: a cancer cell and a normal
cell. The simulated cell junctions in cancer cell are
used to share information with the normal cell. The
goal of the designed in silico experiments was to
find proteins that prevent the formation of gap
junctions and leave the cancer cell uncommunicated.
The early in silico experiments carried out
consisted of running the simulation through a series
of combinations among the concentrations of several
target proteins. e.g., FZD, NDK, DVL, DSH and
APC. After a series of possible arrangements, i.e.,
increasing or decreasing the concentration, or even
removing the protein, we selected two proteins, APC
and NDK.
To determine the necessary concentration of
APC or NKD to prevent the formation of
intercellular junctions, experiments were designed
using concentrations of APC or NDK with the
lowest concentration of 0.001 μM and the highest
concentration of 1000 μM, i.e., 0.001, 0.1, 1, 10, 100
and1000 μM.
We observed that the increase in the
concentration of APC or NKD leads to degradation
of β-catenin in early stages of simulated signal
An in Silico Approach for Understanding the Complex Intercellular Interaction Patterns in Cancer Cells
193

transduction, thus preventing the formation of
intercellular junctions. Regarding APC, at a
concentration of 0.01 μM, it was observed that β-
Catenin disappears at 1,000 milliseconds (ms) and
DVL at 3,000 ms (see Figure 4). Concerning NKD,
at a concentration of 0.001 μM, the disappearance of
β-catenin and DVL is observed at 1000 ms, although
their disappearance is even faster (500 ms) when
using a concentration of 0.01 μM.
4 CONCLUSIONS
The in silico modelling and simulation approach
allowed us to observe, that increasing the
concentration of APC and NKD inhibits the Wnt
signalling pathway, preventing the formation of
intercellular junctions, since the β-CateninCyt is
destroyed and disappears. In silico experimentation
help us to determine the appropriate concentration of
these target molecules.
To carry out the experiments in cancer cells, we
use concentrations of 0.1 μM for both NKD2 and
APC. Using this concentration, β-CateninCyt
quickly disappears, and thus the formation of gap
junctions is avoided. Thanks to in silico experiments
we found two target proteins involved in the
intercellular communication channels and we
observed that their inhibition can stop cancer
development. The next step is to use this result to
guide the in vitro experiments, which in turn will
feedback the in silico modelling and simulation
approach proposed here.
The model of intercellular communication in cancer
cells evolved meaningfully, from the first versions to
later version, after multiple theoretical/experimental
feedbacks which allowed to solve the following
problems that emerged during the execution of the
associated simulation:
1) the earliest models of intercellular
communication in cancer cells did not include all the
required reactions, particularly negative feedback (or
balancing feedback), and 2) the estimated reaction
rate constant of some reactions did not meet the
required value, avoiding that such reactions were
executed at the appropriate time by Gillespie’s
algorithm, resulting that some slower reactions were
executed before the faster reactions.
As part of our future work, 1) we will increase
the size and complexity of the Wnt signalling
network, including other Wnt cross talk pathways,
since if we are able to understand, explore and
control this cellular signalling network, then tissue
invasion and metastasis can be avoided, 2) larger
intercellular communication systems (i.e., 3 o more
cells) will be simulated for verifying how the
proposed approach scales with the system size, and
3) we will use other related simulation tools, such as
MCell (Kerr et al., 2008) and Virtual Cell (Cowan et
al., 2012) for comparison with Cellulat.
Figure 4: APC at a concentration of 0.01 µM. After a series of possible arrangements, we selected two proteins: APC and
NKD. The APC protein is part of the complex that favors the ubiquitination of β-catenin, and therefore its destruction. If
there is no β-catenin, there is no union formation. This is still observed using an APC concentration of 0.001 µM, β -catenin
and DVL disappear around 2500 milliseconds. At this point only the Wnt receptor remains present. On the chart, the x-axis
represents the time in millisecond, and the y-axis represents the concentration of reactants (scaled by 10) in micromolar.
BIOINFORMATICS 2018 - 9th International Conference on Bioinformatics Models, Methods and Algorithms
194

ACKNOWLEDGEMENTS
The authors would like to thank A. Boccacci and O.
Sánchez-Cortés for making a valuable contribution
to this Project.
REFERENCES
Alves, R., Antunes, F., Salvador, A., 2006. Tools for
kinetic modeling of biochemical networks. Nat
Biotechnol. 24(6), 667–72.
Blaheta, R.A., Michaelis, M., Driever, P. H. Cinatl, J. Jr.,
2005. Envolving anticancer drug valproic acid:
Insights into the mechanism and clinical studies. Med
Res Reviews 25(4), 383-397.
Borggrefe, T., Lauth, M., Zwijsen, A., Huylebroeck, D.,
Oswald, F., Giaimo, B.D., 2016. The Notch
intracellular domain integrates signals from Wnt,
Hedgehog, TGFβ/BMP and hypoxia pathways.
Biochim Biophys Acta 1863(2), 303–13.
Brücher, B. L. D. M., Jamall, I.S., 2014. Cell-cell
communication in the tumor microenvironment,
carcinogenesis, and anticancer treatment. Cell Physiol
Biochem. 34, 213–43.
Cárdenas-García, M., González-Pérez, P.P., 2013.
Applying the tuple space-based approach to the
simulation of the caspases, an essential signalling
pathway. J Integr Bioinform. 10(1), 225–34.
Cárdenas-García, M., González-Pérez, P. P., Montagna,
S., Cortés Sánchez, O., Caballero, E. H., 2016.
Modeling Intercellular Communication as a Survival
Strategy of Cancer Cells: An In Silico Approach on a
Flexible Bioinformatics Framework. Bioinformatics
and Biology Insights 10, 5-18.
Ciocchetta, F., Duguid, A., Guerriero, M. L., 2009. A
compartmental model of the cAMP/PKA/MAPK
pathway in bio-PEPA. Proceedings Third Workshop
on Membrane Computing and Biologically Inspired
Process Calculi, MeCBIC 2009, Bologna, Italy,
http://dx.doi.org/10.4204/EPTCS.11.5.
Cowan, A. E., Moraru. I. I, Schaff, J. C., Slepchenko, B.
M., Loew, L. M., 2012. Spatial modeling of cell
signaling networks. Methods Cell Biol. 110, 195–221.
Gelernter, D., 1985. Generative communication in Linda.
ACM Transactions on Programming Languages and
Systems 7(1), 80–112.
Gillespie, D. T., 1977. Exact stochastic simulation of
coupled chemical reactions, The Journal of Physical
Chemistry 81(8), 2340-2361.
González-Pérez, P. P., Cárdenas, M., Camacho, D.,
Franyuti, A., Rosas, O., Lagúnez-Otero, J., 2003.
Cellulat: an agent-based intracellular signalling model.
Biosystems 68(2–3),171–85.
González-Pérez, P. P., Omicini, A., Sbaraglia, M., 2013.
A biochemically inspired coordination-based model
for simulating intracellular signalling pathway. J
Simul. 7(3), 216–26.
González-Pérez, P. P., Cárdenas-García, M., Montagna,
S., 2013. Understanding the PI3K/AKT anti-apoptotic
signalling pathway: a tuple space-based computational
framework for simulating the signal transduction. J
Comput Model. 3(2), 35–65.
Grunsven, V., Vlierberghe, V., 2014. The roles of
transforming growth factor-β, Wnt, Notch and hypoxia
on liver progenitor cells in primary liver tumours
(review). Int J Oncol. 44(4), 1015–22.
Hernández, A. R., Klein, A. M., Kischner, M. W., 2013.
Kinetic responses of b-catenin specify the sites of Wnt
control. Science (338), 1337-1340.
Hoops, S., Sahle, S., Gauges, R., et al., 2006. COPASI: a
complex pathway simulator. Bioinformatics 22(30),
67–74.
Kerr, R. A., Bartol, T. M., Kaminsky, B., et al., 2008. Fast
Monte Carlo simulation methods for biological
reaction-diffusion systems in solution and on surfaces.
SIAM J Sci Comput. 30(31), 26–49.
Lee, E., Salic, A., Kruger, R., Heinrich, R., Kirschner, M.
W., 2003. The roles of APC and Axin derived from
experimental and theoretical analysis of the Wnt
pathways. PLoS Biology 1(1), 116-132.
Lum, L., Chen, C., 2015. Chemical disruption of WNT-
dependent cell fate decision-making mechanisms in
cancer and regenerative medicine. Curr Med Chem.
22(35), 4091–103.
Shimizu, T., Nakagawa, K., 2015. Novel signal
transduction pathways: the molecular basis for
targeted cancer therapies in Hedgehog/Notch/Wnt
pathway. Nihon Rinsho. 73(8), 1342–8.
Swat, M., Thomas, G. L., Belmonte, J. M., Shirinifard, A.,
Hmeljak, D., Glazier, J.A., 2012. Multi-scale
modeling of tissues using CompuCell3D. Methods
Cell Biol. 110, 325–66.
An in Silico Approach for Understanding the Complex Intercellular Interaction Patterns in Cancer Cells
195
