
ASAR Database: An R Tool for Visual Analysis and Storage of
Metagenomes
Askarbek Orakov
1,2
, Nazgul Sakenova
1,2
, Igor Goryanin
1,4,5
and Anatoly Sorokin
3,6
1
Okinawa Institute of Science and Technology, Onna-son, Japan
2
School of Science and Technology, Nazarbayev University, Astana, Kazakhstan
3
Institute of Cell Biophysics RAS, Pushchino, Russia
4
University of Edinburgh, School of Informatics, Edinburgh, U.K.
5
Tianjin Institute of Industrial Biotechnology, Biodesign Centre, Tianjin, China
6
Moscow Institute of Physics and Technology, Dolgoprudny, Russia
Keywords:
Metagenomics, Interactive Data Analysis, Functional Analysis, KEGG Pathways, Taxonomic Analysis.
Abstract:
The functional and taxonomic analysis is the critical step in understanding the interspecies interaction within
the microbial communities. Currently, these types of analysis are run independently, which makes interpreta-
tion of the results hard and error-prone. Here we present A SAR (Advanced metagenomic Sequence Analysis
in R) Database, the interactive tool and the databases for storage and exploratory analysis of the metagenomic
sequencing data along three dimensions: taxonomy, function, and environmental conditions.
1 INTRODUCTION
It is known that 99% of p rokary otic species are not
culturable (Schloss and Handelsman, 2005) either at
all or culture co nditions a re not known. In tha t ci-
rcumstances, the metagenomic analysis becomes an
essential experimental techniq ue for our understan-
ding of composition and functional properties of mi-
crobial communities. In addition to that, decrea-
sing the cost of sequen c ing and increasing throug-
hput of seque ncing machinery cause rapid growth in
availability of th e metagenomic da ta , which makes
the develop ment of tools for functiona l, taxonomic
and metabolic analyses of metagenomes extremely
important (Hugenh oltz and Tyson, 2008; Lindgreen
et al., 2016). Recently the who le genome sequencing
(WGS) become more and mo re popular in compari-
son with 16S, Ribosom al Intergenic Spacer Analysis
(RISA), which compare s the sizes of the intergenic re-
gion between the 16 S rRNA (rrs) and 23 S rDNA (rrl)
genes, and other amplicon sequencing techniques as
it not o nly provides information about the taxonomi-
cal compo sition of the biome but high light its functi-
onal abilities via mapping DNA reads on to pr otein
function database. However, even most promising
current metagenomic analysis tools usually provide
either only taxonomic (Menzel et al., 2 016) or just
functional (Westbrook et al., 2017) a nalysis. Some
tools implement both types of an alysis but indepen-
dently (Keegan et a l., 2016) . That renders data analy-
sis incomplete and leaves a lot of information contai-
ned in the meta genomic datasets undiscovered. Re-
cently we have developed the ASAR (A dvanced met-
agenom ic Sequence Analysis in R) application (Ora-
kov et al., 2017) to fill that gap.
Simultaneous analysis of taxonomic and functio-
nal annotations at the reading level could help answer
many important questions, such as, which taxonom ic
group in a sample is the main contributor to a parti-
cular function or me tabolic pathway. Moreover, abi-
lity to analyze changes in microbiomes in the context
of the metabolic network is the critical requirements
for un derstanding biochemical proce sses in the com-
munity and the presence of competition or symbio-
sis betwee n species. Discovering the most important
metabolic pathways would also considerably improve
the understanding of microbial community evolution.
The core advantage of ASAR is the ab ility to perform
taxonomic and functional analyses simultaneously, by
interactive subsetting and aggregating abundance data
at various levels of taxonomical and functional hier-
archy. It is designed to let researchers drill down to-
wards the most meaningful view of their data in a con-
venient way. It is also possible to perform the compa-
196
Orakov, A., Sakenova, N., Goryanin, I. and Sorokin, A.
ASAR Database: An R Tool for Visual Analysis and Storage of Metagenomes.
DOI: 10.5220/0006722801960200
In Proceedings of the 11th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2018) - Volume 3: BIOINFORMATICS, pages 196-200
ISBN: 978-989-758-280-6
Copyright © 2018 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

rative analysis of the KEGG metabolic pathways (Ka-
nehisa et al., 2016), by exploring the pathways enri-
chment and visualizing the pa thways themselves.
Original ASAR application was designed to deal
with data in me mory. It is not uncommon in metage-
nomics to have the repetitive collection of samples as
a time series. In this case, application sometimes has
to dea l with datasets of hundr e ds of samples, which
do n ot fit into the memory of regular workstation. To
handle large datasets, we aug mented the ASAR appli-
cation with the database to store r aw data and perform
the aggregation and selection.
2 METHODS
The application was written in R programming lan-
guage (R Core Team, 2017) and Shiny platform
(Chang et al., 2017) was used to make it web-
based and user-interactive. Thanks to R and Shiny,
the ap plication can both be used locally at ma chi-
nes with installed R and as web-service. MonetDB
(https://www.monetdb.org/) was used as DBMS and
MonetDB.R (Muehleisen et al., 2017) pac kage was
used for connection between R application and
DBMS. The application requires following R packa-
ges: dplyr (Wickham et al., 2017) and data .table (Do-
wle and Srinivasan, 2017) for efficient data mani-
pulation; ggplot2 (Wickham, 2016), gp lots ( Warnes
et a l., 2016), RColorBrewer (Neuwirth, 2014) and
d3heatmap (Cheng and Galili, 2016) for visualiza-
tion; pa thview (Luo et al., 201 3) and png (Urbanek,
2013) for exportin g the results.
Two datasets were used for development of the
application. The sm a ll dataset contains 1 1 me tage-
nomes (total size 45 GB) from swine waste microbial
fuel cell (MFC) performance analysis proje c t (Khi-
lyas et al., 2017). Th e moderate dataset consists of
172 metagenomes (total size 195 GB) from longitu-
dinal monitoring of the MFC wastewater treatment of
Spent Wash (Dimou et al., 2014). Both datasets were
loaded into the database separa te ly. The small d a ta set
was used for the performance comparison with the in-
memory application. The mod erate da taset does not
fit into memory, so it was used for demonstration of
the performance of the database version of the app.
3 RESULTS
3.1 The WGS Data
Sequencin g data usually comes as a set of short DNA
reads, which are mapped to genomic and functional
databases for annotation by tools like Kaiju (Menzel
et al., 2016), Paladin (Westbrook et al., 2017), and
MG-RAST (Keegan et al., 2016). After joining of
taxonomical and functional annotation, the data form
2D matrix with species in rows and functions in co-
lumns. In that matrix, each cell contains the abun-
dances of reads mapped to the particular function in
particular species. Analysis of single metagenom e is
quite rare, usually, metagenomes obtained at several
sets of environmental conditions, time points and per-
turbations are analyzed. Th at set of samples forms the
third dimension of the dataset.
The analysis of multidimensional datasets is a
tricky task; this is one reason why people usually
analyze taxono my and functional data separately: ag-
gregation along f unctional or taxonomic dimension
forms the 2D matrix from the data, which is more
straightfor ward for visualization and inte rpretation.
The similar type of task was solved in business ana-
lytics in the middle of 80s by development concept
of the data cube (Kimball and Ross, 2011). In our
case, the data cube is the 3D array with taxo nomy,
function, and metagenome as dime nsions and read
counts as cell content. Elements of two of dimensi-
ons form hierarchies: taxono mic and f unctional. The
components of metagenome dimension usually orga-
nized into kind of d esign matrix either explicitly by
planning experiment upfront, or implicitly by explo-
ring the spatial and te mporal variability o f a microbial
community under investigation.
We de sig ned ASAR (Orakov et al., 2 017) appli-
cation for inte ractive analysis of the whole dataset by
application aggregation and selection operations dyn -
amically and exploration of the obtained 2D matrices
visually. At the moment we are using the annotation
files generated by MG- RAST pipeline, but any other
annotation pipeline, which assigns annotation at the
DNA read level, such as Kaiju, Paladin, QIIME, etc.,
would give similar resu lts. The MG-RAST was cho-
sen because its annotation is based o n the common
database a nd so self-consistent. The pr ocedure of im-
port other types of data is the same, but mismatches
caused by use different references during DNA read
annotation won’t be fixed.
3.1.1 Selection and Aggregation
Interactive a pplication of Selection and Aggregation
is the essential steps of dynamic exploration of the
3D data cube. Selection operation allows the user
to navigate thro ugh hierarchy by selecting element at
some high e r level of the tree and analyze the sub set
of the cube underneath p art chosen. For example, the
user can choose Deltaproteobacteria at the class level
of taxonomy a nd restrict consideration to species and
ASAR Database: An R Tool for Visual Analysis and Storage of Metagenomes
197
ID int4
level int4
usp varchar(2055)
species varchar(2055)
genus varchar(2055)
family varchar(2055)
order varchar(2055)
class varchar(2055)
phylum varchar(2055)
domain varchar(2055)
name varchar(2055)
asar.taxonomy
ID int4
FUN1 varchar(4055)
FUN2 varchar(4055)
FUN3 varchar(4055)
FUN4 varchar(4055)
asar.seed
id int4
access varchar(25)
function varchar(4055)
asar.KO
ID int4
access varchar(25)
name varchar(4055)
description varchar(8255)
asar.kegg_pathway
id int4
mg.id int4
read.id varchar(70)
md5sum char(32)
identity float4
al.length int4
n.mis int4
n.gap int4
q.start int4
q.end int4
h.start int4
h.end int4
e.val float4
score float4
asar.reads
md5sum char(32)
asar.annot
ID int4
mgrast.id varchar(255)
name varchar(2055)
description varchar(8255)
ProjectID int4
asar.metagenome
metagenomeID int4
name varchar(255)
value varchar(255)
description varchar(8255)
asar.metadata
kegg.pathwayID int4
KOid int4
asar.kegg_pathway_KO
Powered ByVisual Paradigm Community Edition
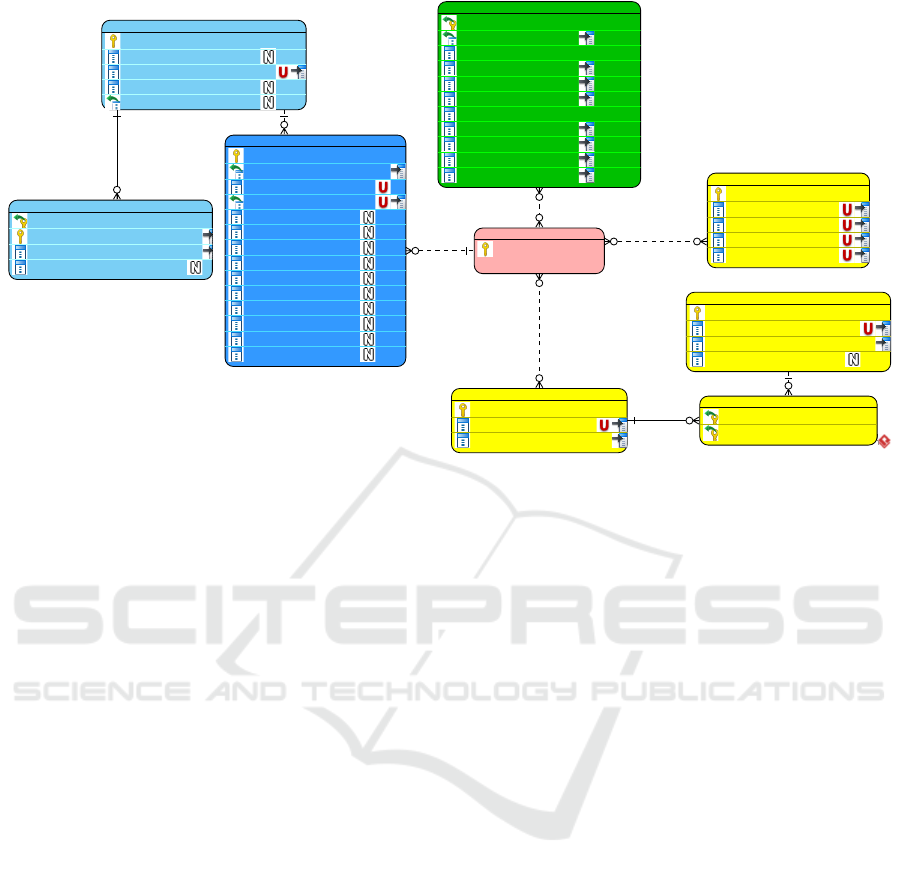
Figure 1: The database structure diagram. The fact table is shown in blue, functional annotation shown in yellow, taxonomic
annotation shown in green. ‘U’ icon indicates t he columns with the unique constraint. ‘N’ icon indicates the nullable columns.
Columns included in the primary key are marked by the key symbol.
functions in that class only. A ggregation operation al-
lows the u ser to summariz e the data at some level of
the hierarchy, by summing up the content of cells cor-
respond ing to the same element at selected Aggrega-
tion level. For example , the reliability of data at strain
level is usually low, so it is common to Aggregate the
data up to the genus level.
The application of Selec tion and Aggregation to
the metagenome axis, by choosing the particular field
in the metada ta and use it as a hierarchy level fo r se-
lection and aggregation. For example, ana lysis of mi-
crobial fuel cell (MFC) microbiome usually consider
anodic biofilm separately from the planktonic com-
munity, so we can choose “Part of MFC” field from
metadata and Select “Anode ” value to study the com-
position of anodic biofilm only. We can also aggre-
gate all planktonic communities into one matrix and
explore their dependence on tim e or initial commu-
nity composition.
3.1.2 SEED Annotation Analysis
Shiny Application has five tabs, four of w hich ar e he-
atmaps and last is the KEGG pathway dia gram (Fi-
gure 2).
First three heatmaps a re three different projecti-
ons of the 3D dataset: function vs. taxonomy (F/T),
function vs. metagenome (F/M) and taxonom y vs.
metagenome (T/M). So, in the first heatmap, you
can see the a bunda nce of each intersection between
function and taxon in a single metag e nome samp le.
The next two heatmaps represent traditional fun ctio-
nal and taxonomic ana lysis and a llow to compare en-
richments of functions or taxons among selected met-
agenom es. For both function s and taxons u ser can
choose pa rticular level and value to work with a nd
the level at which all data will be aggregated. The
taxonomic hierarchy levels are taken from MG-RAST
(Keegan et al., 2016) and SEED hierarchy (Overbeek
et al., 2013) is used for functio ns.
3.1.3 KEGG Annotation Analysis
In the fourth heatmap one can compare KEGG
pathway (Kanehisa et al., 2016) enrichments in ord er
of the descending value of standard deviation among
selected samples in a selected taxon. After one finds
the pathway of interest, choosing the pathway name
in the last tab will draw its KEGG diagram. In the
diagram, every enzyme will correspond to a rectangle
where samples are colored according to values of their
contribution to the abundance of that enzym e in the
community.
3.2 Database Structure
The structure of the database (Figur e 1) follows stan -
dard Online a nalytical processing (OLAP) Snowflake
pattern (Ponniah, 2010) with asar.reads as the fact ta-
ble. The icons on the diagram follow The metage-
nome and annot tables define two main dimensions.
BIOINFORMATICS 2018 - 9th International Conference on Bioinformatics Models, Methods and Algorithms
198
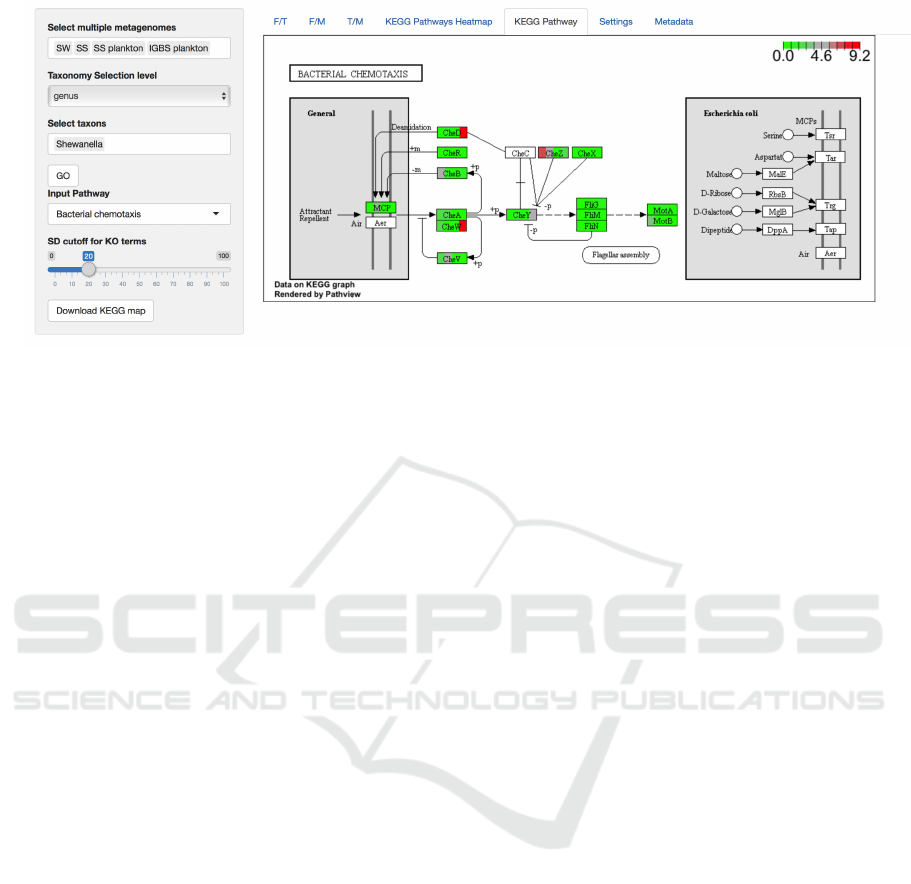
Figure 2: The KEGG diagram visualization. The selection panel is on the left. It i s possible to choose metagenomes of
interest, taxonomy selection l evel and value and t he pathway to show. The “GO” button prevents unintended drawing of the
pathway, which requires access to the KEGG database and is time-consuming.
The former one for non-hierarchical sample dimen-
sion, the latter for both hie rarchical taxo nomic (taxo-
nomy table) and functional (function table) dimensi-
ons. The ko ta ble, another connection of the annot
table, provides KEGG annotation for pathway analy-
sis.
All selection and aggregation operations along
taxonomy and function dimensions of the d a ta cube
are performed in memory in the same way as in origi-
nal ASAR application, while for selection and aggre-
gation operations along metagenomic dimension the
SQL queries to the database is used. That way o f in-
teraction with the database was chosen to reduce the
response time of the application, as the manipu lation
along the functional and taxonomic dimensions are
much more often compare to m odification of selection
and aggregation criteria along samples dimension.
The taxonomic hierarchy levels are taken fro m
MG-RAST (Keegan e t al., 2016 ). It consists of eight
levels from domain to strain levels. The read could be
assigned to any level of the taxonomy, so ”least com-
mon ancestor” annotation method could be used. The
structure of functional annotation follows the SEED
hierarchy (Overbeek et al., 2 013) that consists of four
levels. Level 1 of the SEED hierarchy correspon ds to
individual enzyme functions, while major functional
groups like “DNA metabolism” or “Virulence” form
the level 4 of the tree. Specific kind of annotation is
KEGG orthology, which is required for m apping of
metagenomic data onto th e KE G G pathway.
4 CONCLUSIONS
Our post-anno ta tion analysis and visualization tool
uses data integration algorithm to merge taxonomic
and functional data annotated at the DNA read le-
vel. The resulting 3D dataset with axes of Functions,
Taxonomy and Metage nome samples is visualized via
three heatmaps of each axis versus two others (F/T,
F/M, T/M). Additio nally, KEGG pathway enrichmen t
sorting/heatmap and its map visualization a re imple-
mented.
We have tested the performance of the database
on I ntel Core i5, 32 GB RAM workstation with sm all
(11 metagenomes, total size 45 GB) and modera te
(172 metagenom es, total size 195 GB) datasets. The
average response time was in a range of 10 sec for in-
memory data tr ansformation and up to 2 min for DB
SQL query. The database upload time was 5 minu-
tes for the small da ta set and 10 minutes for the mo-
derate one. The source code of ASAR is free and
accessible at GitHub (https://github.com/Askarbek-
orakov/ASAR).
ACKNOWLEDGEMENTS
Members of OIST BSU UNIT, Irina Kh ilyas for pro-
viding d a ta , OIST NGS Sectio n for sequencing servi-
ces. Dr. Jeannette Kunz for suppor ting internship.
This work has been supported by the OIST funding.
REFERENCES
Chang, W., Cheng, J., Allaire, J., Xie, Y., and McPherson,
J. (2017). shiny: Web Application Framework for R.
Cheng, J. and Galili, T. (2016). d3heatmap: Interactive
Heat Maps Using ’htmlwi dgets’ and ’D3.js’. R
package version 0.6.1.1.
ASAR Database: An R Tool for Visual Analysis and Storage of Metagenomes
199

Dimou, O., Andresen, J., Feodorovich, V., Goryanin, I.,
Harper, A ., and Simpson, D. (2014). Optimisation of
scale-up of microbial fuel cell for sustainable waste-
water treatment with positive net energy generation.
New Biotechnology, 31:S213.
Dowle, M. and Srinivasan, A. (2017). data.table: Extension
of ‘data.frame‘. R package version 1.10.4-3.
Hugenholtz, P. and Tyson, G. W. ( 2008). Microbiology:
metagenomics. Nature, 455(7212):481–483.
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and
Tanabe, M. (2016). KEGG as a reference resource for
gene and protein annotation. Nucleic Acids Research,
44(D1):D457–62.
Keegan, K. P., Glass, E. M., and Meyer, F. (2016). MG-
RAST, a Metagenomics Service for Analysis of Mi-
crobial Community Structure and Function. Methods
in Molecular Biology (Clifton, N.J.), 1399:207–233.
Khilyas, I. V., S orokin, A., Kiseleva, L., Simpson, D.
J. W., Fedorovich, V., Sharipova, M. R., Kainuma,
M., Cohen, M. F., and Goryanin, I. (2017). Compa-
rative Metagenomic Analysis of E lectrogenic Micro-
bial Communities in Differentially Inoculated Swine
Wastewater-Fed Microbial Fuel Cells. Scientifica,
2017(5-6):1–10.
Kimball, R. and Ross, M. (2011). The Data Warehouse
Toolkit. The Complete Guide to Dimensional Mo-
deling. John Wiley & Sons.
Lindgreen, S., Adair, K. L., and Gardner, P. P. (2016). An
evaluation of the accuracy and speed of metagenome
analysis tools. Scientific Reports, 6(1):19233.
Luo, Weijun, Brouwer, and Cory (2013). Pat-
hview: an r /bioconductor package for pathway-based
data integration and visualization. Bioinformatics,
29(14):1830–1831.
Menzel, P., Ng, K. L., and K r ogh, A. (2016). Fast and sen-
sitive taxonomic classification for metagenomics with
Kaiju. Nature Communications, 7:11257.
Muehleisen, H., Damico, A., Raasveldt, M., Lumley, T.,
and Team, M. D. (2017). MonetDBLite: In-Process
Version of MonetDB for R. MonetDB. R package
version 0.4.1.
Neuwirth, E. (2014). RColorBrewer: ColorBrewer Palettes.
R package version 1.1-2.
Orakov, A., Sakenova, N., Sorokin, A., and Goryanin, I.
(2017). ASAR: visual analysis of metagenomes in R.
OIST, Okinawa, Japan.
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis,
J. J., Disz, T., Edwards, R . A., Gerdes, S ., Parrello, B.,
Shukla, M., Vonstein, V., Wattam, A. R., Xia, F., and
Stevens, R. (2013). The SEED and the Rapid Annota-
tion of microbial genomes using Subsystems Techno-
logy (RAST). Nucleic Acids Research, 42(D1):D206–
D214.
Ponniah, P. (2010). Olap in the Datawarehouse, pages 373–
406. John Wiley & Sons, Inc.
R Core Team (2017). R: A Language and Environment for
Statistical Computing. R Foundation for Statistical
Computing, Vienna, Austria.
Schloss, P. D. and Handelsman, J. (2005). Metagenomics
for studying unculturable microorganisms: cutting the
Gordian knot. Genome Biology, 6(8):229.
Urbanek, S. (2013). png: Read and write PNG images. R
package version 0.1-7.
Warnes, G. R. , Bolker, B., Bonebakker, L., Gentleman, R.,
Liaw, W. H. A., Lumley, T., Maechler, M., Magnus-
son, A., Moeller, S., S chwartz, M., and Venables, B.
(2016). gplots: Various R Programming Tools for
Plotting Data. R package version 3.0.1.
Westbrook, A., Ramsdell, J., Schuelke, T., Normington,
L., Bergeron, R. D., Thomas, W. K., and MacMa-
nes, M. D. (2017). PALADIN: protein alignment
for functional profiling whole metagenome shotgun
data. Bioinformatics (Oxford, England), 33(10):1473–
1478.
Wickham, H. (2016). ggplot2: Elegant Graphics for Data
Analysis. Springer-Verlag New York.
Wickham, H., Francois, R., Henry, L., and M¨uller, K.
(2017). dplyr: A Grammar of Data Manipulation. R
package version 0.7.4.
BIOINFORMATICS 2018 - 9th International Conference on Bioinformatics Models, Methods and Algorithms
200
