
First-principles Study on LiFePO
4
Materials for Lithium-ion
Battery
S Z Wang
1,2,3,*
, G Zhang
2
, J L Gao
3
, J Wang
2
, Y Y Wang
2
,C J Nan
1
,
D Y Huang
1
, L L Chen
1
,J F Song
1
and P H Luo
1
1
Nanyang Institute of Technology, Henan, P.R. China
2
Nanyang Explosion Protected Electrical Apparatus Research Institute, Henan, P.R.
China
3
Henan Polytechnic University, Henan, P.R. China
Corresponding author’s e-mail: S Z Wang, 0610103017@tongji.edu.cn,
szwang0001@163.com
Abstract. In this paper, the band structure and density of states of LiFePO
4
are calculated by
first principles. It is found that LiFePO
4
is a semiconductor with the f band gap of 0.786eV.
The electrochemical properties of LiFePO
4
are mainly influenced by Fe element. The
thermodynamic properties of cathode material LiFePO
4
for lithium ion battery were also
studied. The entropy S, the specific heat capacity C and the enthalpy H of the LiFePO
4
increased with the increase of temperature and the Gibbs free energy G decreased with the
increase of temperature in the paper.
1. Introduction
With the increasing demand of portable electronic products, rechargeable electric vehicles and other
traffic equipment, lithium ion batteries have become the most widely used power batteries at present.
It is also a hot spot in the research and industry all over the world. Compared with other cathode
materials for lithium ion batteries, LiFePO4 has high theoretical capacity, good cycling performance,
stable performance and abundant raw materials. Moreover, LiFePO4 become one of the first choices
of lithium-ion power battery materials because of its Low cost, environmental protection and other
advantages [1-4].
In order to improve the utilization of LiFePO
4
material, it is necessary to improve the
electrochemical performance of this material. Researchers have done a lot of work in experiments
[5-9]. However, the theoretical research of LiFePO
4
is also very important [10]. Therefore, we focus
on the properties of LiFePO
4
electronic structure, so as to understand the electronic properties and
some chemical bonds of LiFePO
4
materials in this paper. Furthermore, there is important guiding
significance for improving the conductivity of and ion diffusivity of LiFePO
4
materials. The results
of other researchers provide the conditions and basis for us to understand LiFePO
4
theoretically.
Moreover, it can help us improve the performance of LiFePO
4
in theory. Recently, the first principle
calculation method combined with molecular dynamics has made great contributions to the design
synthetic simulation and performance evaluation of materials. And first principle calculation method
Wang, S., Zhang, G., Gao, J., Wang, J., Wang, Y., Nan, C., Huang, D., Chen, L., Song, J. and Luo, P.
First-principles Study on LiFePO4 Materials for Lithium-Ion Battery.
In Proceedings of the International Workshop on Materials, Chemistry and Engineering (IWMCE 2018), pages 133-138
ISBN: 978-989-758-346-9
Copyright © 2018 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
133
has become the core technology of the science of material computing. In this paper, we use the first
principles to study LiFePO
4
materials from solid state physics.
2. Simulation and calculation
2.1. Structures
LiFePO
4
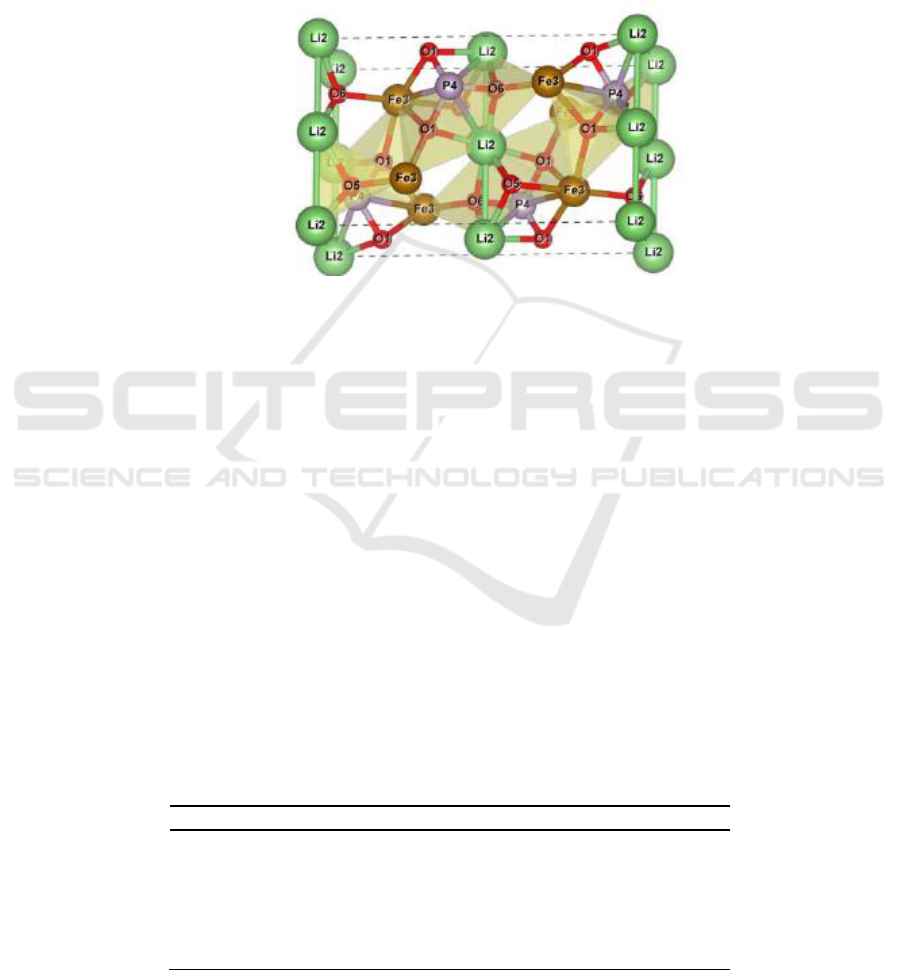
exists mainly in the form of lithium iron phosphate in nature. Its stereoscopic diagram is
shown as shown in Figure 1. The crystal structure of LiFePO
4
is olivine type, and it has good
regularity. And it is easy to form highly ordered lattice.
Figure 1. Stereoscopic diagram of LiFePO
4.
As shown in Figure 1, LiFePO
4
crystal belongs to the Pnma orthogonal space group. The oxygen
atoms accumulate densely close to the six parties. And Fe and Li are respectively located 4C and 4A
bits in octahedron center of O atom. This leads to the formation of FeO
6
octahedron and LiO
6
octahedron. P atom is located 4C bits in the central position of O atom tetrahedron. In this way, the
PO
4
tetrahedron is formed. Li
+
parallel to the c axis forms a continuous linear chain in t 4aposition.
And Li+ moved along the c axis in two-dimensional diffusion. It can embed in charge and discharge
process freely. Phosphoric acid has the function of supporting the whole material frame. It makes the
material has good thermal stability and cycling performance.
The charge discharge process is carried out between LiFePO
4
and FePO
4
, and the cell parameters
of LiFePO
4
are a=10.6380 Å, b=5.9630 Å, c=4.5280 Å. And the volume changes 6.8% during the
charge discharge process. At the same time, in the process of lithium ion deintercalation, the crystal
structure is not rearranged. It maintains olivine structure. Therefore, LiFePO
4
has excellent cycle
performance.
2.2. Structure optimization
The coordinate parameters and are shown in Table 1.
Table 1. Lattice parameters (SpaceGroup: Pnma, SG Number: 63, Crvst Sys: orthorhombic).
atom
x
y
z
1
Li
0
0
0
2
Fe
0.2820
0.250
0.9734
3
P
0.0951
0.250
0.4187
4
O1
0.0922
0.250
0.744
5
O2
0.4547
0.250
0.211
6
O3
0.1626
0.0477
0.2844
IWMCE 2018 - International Workshop on Materials, Chemistry and Engineering
134
After geometric optimization, the most stable structure of LiFePO
4
can be obtained. The
parameters of LiFePO
4
crystal after optimization are shown in Table 2. The cell volume increased
from 287.231 Å
3
to 267.211 Å
3
.
Table 2. Lattice parameters before and after optimization.
a(Å)
B(Å)
C(Å)
Pre
optimization
After optimization
Pre
optimization
After
optimization
Pre
optimization
After
optimization
10.6380
9.8856
5.9630
5.7932
4.5280
4.6658
3. Results and analysis
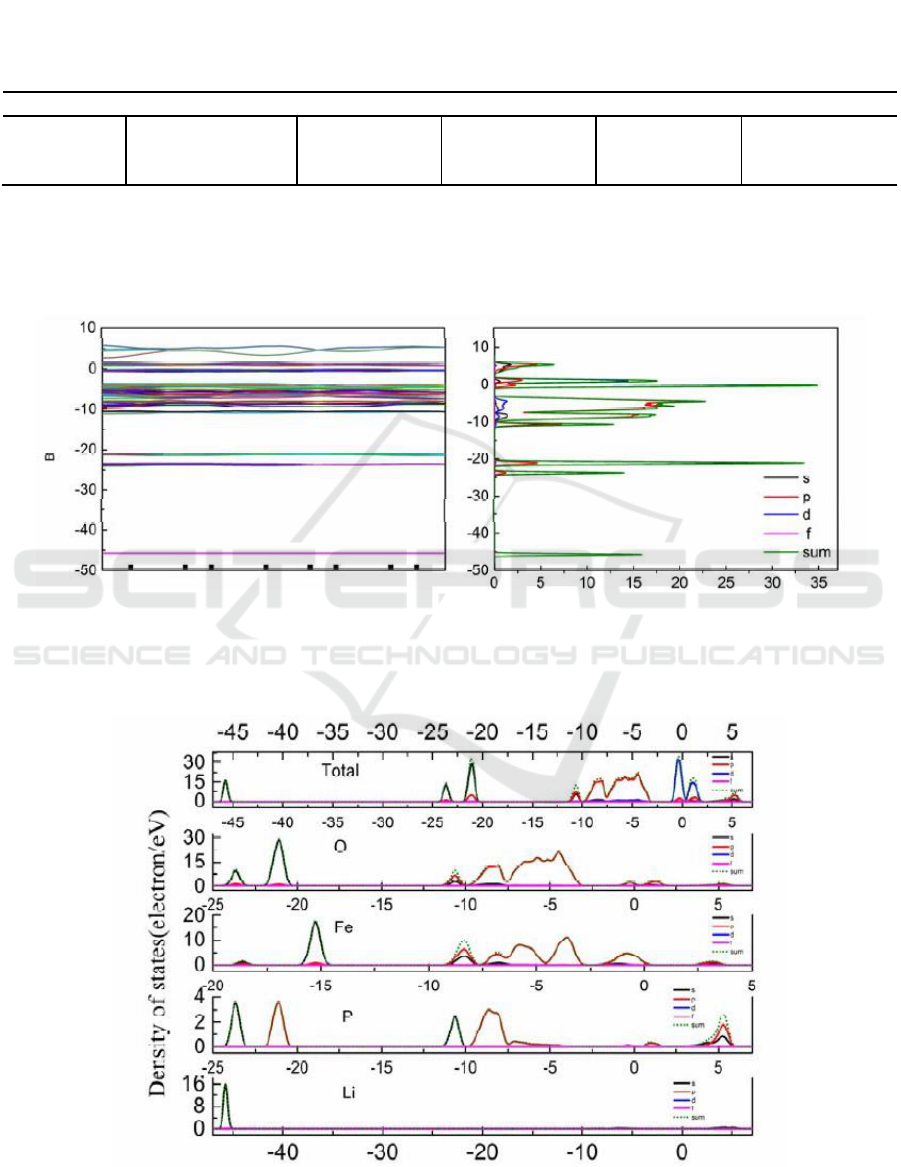
The band structure and the density states of cathode material LiFePO
4
for lithium ion batteries are
calculated. As seen in figures 2 and 3, LiFePO
4
shows the characteristics of semiconductors. And the
band gap of is 0.768eV, which is greater than reports of S.Q. Shi [11-12].
Figure 2. Total Energy band and Density of States of LiFePO
4.
The valence band is mainly composed of the electron on p orbit of O and d orbit of Fe below
Fermi surface. FeO
6
octahedron is formed at the same time. The conduction band above Fermi
surface is mainly contributed of electrons on d orbital of Fe.
Figure 3. Density of States of Li atoms, P atoms, O atoms, Fe atoms.
First-principles Study on LiFePO4 Materials for Lithium-Ion Battery
135

Figure 3 indicates that the electrochemical properties of LiFePO
4
are mainly affected by Fe atom.
The contribution of electron on s orbital of Li near Fermi level is very small. The conduction band
between 2.5eVand 7eV is mainly contributed by the electron on p orbital of Li and electron on p orbit
of Fe. But its effect is very weak, which mainly affects the conductivity of LiFePO
4
. But the effect is
Table 3. The thermodynamic properties of LiFePO
4.
Temperature
(T)
Entropy
(J· mol
-1
·K
-1
)
Specific heat capacity
(J· mol
-1
·K
-1
)
Enthalpy
(kJ· mol
-1
)
Gibbs free energy
(kJ·mol
-1
)
100
55.671
128.7426
0.25485
0.24928
125
90.3546
183.6702
0.25876
0.24746
150
128.394
234.2802
0.264
0.24473
175
167.958
279.2034
0.27043
0.24103
200
207.8664
318.4902
0.27791
0.23633
225
247.401
352.7622
0.28631
0.23064
250
286.1544
382.7628
0.29551
0.22397
275
323.904
409.1766
0.30542
0.21634
300
360.528
432.5538
0.31594
0.20779
325
395.9886
453.3354
0.32702
0.19833
350
430.2732
471.8742
0.33859
0.188
375
463.407
488.4684
0.3506
0.17682
400
495.4152
503.349
0.363
0.16483
425
526.3398
516.726
0.37575
0.15206
450
556.2228
528.7674
0.38883
0.13853
475
585.1062
539.6328
0.40218
0.12426
500
613.0404
549.4482
0.4158
0.10928
525
640.0674
558.3396
0.42965
0.09361
550
666.2292
566.3994
0.44371
0.07728
575
691.572
573.7242
0.45796
0.06031
600
716.1294
580.3938
0.47239
0.04271
625
739.9476
586.4754
0.48698
0.02451
650
763.0602
592.0278
0.50171
0.00572
675
785.5008
597.114
0.51657
-0.01364
700
807.303
601.776
0.53156
-0.03355
725
828.4962
606.06
0.54666
-0.054
750
849.1098
610.0038
0.56186
-0.07497
775
869.169
613.6368
0.57716
-0.09645
800
888.7074
616.9884
0.59254
-0.11842
825
907.7418
620.0922
0.608
-0.14088
850
926.2932
622.965
0.62354
-0.16381
875
944.391
625.632
0.63915
-0.18719
900
962.052
628.11
0.65482
-0.21102
925
979.293
630.4158
0.67056
-0.23529
950
996.135
632.562
0.68634
-0.25998
975
1012.5906
634.5696
0.70218
-0.2851
1000
1028.6808
636.4428
0.71807
-0.31061
IWMCE 2018 - International Workshop on Materials, Chemistry and Engineering
136

very weak, which mainly affects the conductivity of LiFePO
4
. The low energy level lies between -40
and -45eV, mainly composed of electron on s orbit of Li atom.
The thermodynamic properties are calculated at 1 atmospheric pressure. The thermodynamic
temperature is 100 to 1000K. And it is measured once each interval of 25K. Thermodynamic data of
LiFePO
4
is shown in Table 3. The Gibbs free energy G, entropy S, heat capability C and enthalpy H
of LiFePO
4
changed with temperature respectively according to the calculation results shown in
Table 3.
At the same time, Entropy S and enthalpy H of LiFePO
4
increase with the increase of temperature,
while Gibbs free energy G decreases with the increase of temperature, which is in accordance with
thermodynamic law. Gibbs free energy of LiFePO
4
decrease slowly at the beginning. After 250K,
Gibbs's free energy decreased linearly with the increase of temperature. The entropy of LiFePO
4
increases linearly with temperature. The enthalpy of LiFePO
4
changed slowly at the beginning stage,
and the enthalpy increased rapidly with temperature after 300K. The heat capability increases fast
with temperature before 300K. And the heat capability increases with temperature slowly after 300K.
4. Conclusions
In this paper, the electronic structure and thermodynamic properties of LiFePO
4
for lithium ion
batteries cathode materials were calculated by first principles calculations based on density functional
theory. LiFePO
4
exhibits the characteristics of semiconductors by calculating the band structure and
density states of LiFePO
4
. The entropy S and enthalpy H of LiFePO
4
increase with the increase of
temperature, while the Gibbs free energy G decreases with the increase of temperature, which is
consistent with the thermodynamic law. The microstructure and thermodynamic parameters of
lithium ion battery cathode material LiFePO
4
obtained in the paper can provide theoretical guidance
for the practical application of lithium ion batteries.
Acknowledgement
The authors gratefully acknowledge the financial supported by Research of the basic and advanced
technology of Henan Province (No.162300410069, 172400410319), National Natural Science
Foundation of China (Grant No. 61371058), and Key scientific research projects of Henan Province
(No.16A140046), Research project of Nanyang Institute of Technology (SFX201808, HXCK2016018,
NIT2017JY-119,50104033).
References
[1] Braun P V, Cho J, Pikul J H, King W P and Zhang H 2012 Curr. Opin. Solid State Mater. Sci.
16 186
[2] Scrosati B, Carche J. 2010 J. Power Sources 195 2419.
[3] Wang Q S, Ping P, Zhao X J, Chu G Q, Sun J H and Chen C H 2012 J. Power Sources 208
210
[4] Lisbona D and Snee T 2011 process saf environ 89 434-442
[5] Wang K, Hissel D, Péra M C, Steiner N, Marra D, Sorrentino M, Pianese C, Monteverde M,
Cardone P and Saarinen J 2011 Int. J. Hydrogen Energy 36 7212
[6] Scott I D, Jung Y S, Cavanagh A S, Yan Y, Dillon A C, George S M and Lee S H 2011 Nano
Lett. 11 414
[7] Okubo M, Hosono E , Kim J, Enomoto M , Kojima N, Kudo T, Zhou H and Honma I 2007 J.
Am. Chem. Soc. 129 7444
[8] Yang S Y , Wang X Y , Chen Q Q , Yang X K , Li J J and Wei Q L 2012 J. Solid State
Electrochem. 16 481
[9] Hariprakash B, Gaffoor S A and Shukla AK 2009 J. Power Sources 191 149
[10] Xu Y N, Ching W Y and Chiang Y M 2004 J. Appl. Phys. 95 6583.
First-principles Study on LiFePO4 Materials for Lithium-Ion Battery
137

[11] Shi S Q, Ouyang C Y, Xiong Z H, Liu L J, Wang Z X, Li H, Wang D S, Chen L Q and Huang
X J 2005 Phys. Rev. B 71 144404
[12] Xu J and Chen G 2010 Physica B 405 803
IWMCE 2018 - International Workshop on Materials, Chemistry and Engineering
138
