
Artificial Neural Network Approach to Prediction of Protein-RNA
Residue-base Contacts
Morihiro Hayashida
1
, Jose Nacher
2
and Hitoshi Koyano
3
1
Department of Electrical Engineering and Computer Science, National Institute of Technology,
Matsue College, Matsue, Shimane, Japan
2
Department of Information Science, Faculty of Science, Toho University, Funabashi, Chiba, Japan
3
School of Life Science and Technology, Tokyo Institute of Technology, Meguro-ku, Tokyo, Japan
Keywords:
Fully Connected Neural Network, Protein-RNA Interaction, Residue-base Contact.
Abstract:
Protein-RNA complexes play essential roles in a cell, and are involved in the post-transcriptional regulation of
gene expression. Therefore, it is important to analyze and elucidate structures of protein-RNA complexes and
also contacts between residues and bases in their interactions. A method based on conditional random fields
(CRFs) was developed for predicting residue-base contacts using evolutionary relationships between individ-
ual positions of a residue and a base. Further, the probabilistic model was modified to improve the prediction
accuracy. Recently, many researchers focus on deep neural networks due to its classification performance. In
this paper, we develop a neural network with five layers for predicting residue-base contacts. From computa-
tional experiments, in terms of the area under the receiver operating characteristic curve (AUC), the predictive
performance of our proposed method was comparable or better than those of the CRF-based methods.
1 INTRODUCTION
Interactions between proteins and RNAs are involved
in the post-transcriptional regulation of gene expres-
sion including alternative splicing, polyadenylation,
localization and translation (Glisovic et al., 2008).
For instance, a ribosome is formed with multiple pro-
teins and RNAs, and synthesizes proteins from mes-
senger RNAs. Disruption of protein-RNA interac-
tions may cause various diseases including cancers.
Therefore, it is needed to obtain precise knowledge of
contact positions between proteins and RNAs to un-
derstand their molecular function.
It is known that there are several RNA-binding
domains such as RNA recognition motif (RRM),
heterogeneous nuclear ribonucleoproteins (hnRNPs)
(Beyer et al., 1977), the K-homology domains (Siomi
et al., 1993), double-stranded RNA-binding domains
(dsRBD) (Feng et al., 1992), TIA-1 (Kedersha et al.,
1999) and zinc fingers (Hall, 2005). The sequence
and structural properties of RNA-protein interaction
sites in 211 RNA-protein chain pairs were investi-
gated, and it was reported that 78% of hydrogen bonds
involve amino acid side chains, and the remaining
involve the protein backbone (Gupta and Gribskov,
2011). Several computational methods have been
developed for predicting RNA-binding amino acid
residues in proteins. Sun et al. proposed RNAProSite,
which utilizes the random forest (Breiman, 2001)
with electrostatic feature, triplet interface propensity,
position-specific scoring matrices (PSSM) profile, ge-
ometrical characteristic and physicochemical prop-
erty (Sun et al., 2016). Sharan et al. developed an
integrated pipeline, called APRICOT, that identifies
functional motifs in protein sequences using PSSMs,
hidden Markov models of RNA-binding domains, and
sequence-based features (Sharan et al., 2017). It was
reported that APRICOT achieved sensitivities higher
than or as good as RNAProSite and other high per-
forming predictors. Tang et al. proposed PredRBR
that utilizes gradient tree boosting and many kinds
of sequence and structural site features (Tang et al.,
2017).
On the other hand, concerning binding sites of
RNAs, 24.6% of hydrogen bonds involve nucleobase-
specific interactions, and the remaining involve the
RNA backbone (Gupta and Gribskov, 2011). Ali-
panahi et al. proposed DeepBind that uses many
RNA sequences with binding scores determined from
protein binding microarray experiments, and extracts
motif features by a deep learning technique, which
outperformed 26 other methods such as FeatureRE-
Hayashida, M., Nacher, J. and Koyano, H.
Artificial Neural Network Approach to Prediction of Protein-RNA Residue-base Contacts.
DOI: 10.5220/0007348101630167
In Proceedings of the 12th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2019), pages 163-167
ISBN: 978-989-758-353-7
Copyright
c
2019 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
163

DUCE (Weirauch et al., 2013) and BEEML-PBM
(Zhao et al., 2011) when the correlation between the
predicted and actual probe intensities was evaluated
(Alipanahi et al., 2015). Zeng et al. systematically
examined various models of convolutional neural net-
works to further improve DeepBind, and observed
that deploying more convolutional kernels was always
beneficial, but the local-pooling and additional con-
volutional layers were useful only in the motif oc-
cupancy task when higher-order features existed, and
they did not achieve significant improvement (Zeng
et al., 2016). These methods were developed for iden-
tifying RNA and DNA motifs bounded by transcrip-
tion factors. However, both positions in binding RNA
and protein were not identified. Methods for pre-
dicting residue-base contacts in protein-RNA interac-
tions were proposed using conditional random fields
(CRFs), which used evolutionary measurements ob-
tained from multiple sequence alignments (Hayashida
et al., 2013; Hayashida et al., 2018). For analyzing
mechanisms of protein-RNA interactions in detail, we
also deal with the problem of predicting residue-base
contacts.
Recently, artificial neural network techniques (Le-
Cun et al., 2015; Glorot et al., 2011) have been ap-
plied in many research fields due to its high classifica-
tion performance. We explore neural network archi-
tectures to overcome the prediction accuracy by the
CRF-based method. For evaluating the proposed neu-
ral network, we perform computational experiments
as done in the previous study. The results show that
in terms of the area under the receiver operating char-
acteristic curve (AUC), the predictive performance of
our proposed method was comparable or better than
those of the CRF-based methods.
2 METHODS
We address the following problem. Given two se-
quences p
p
p = p
1
··· p
|p
p
p|
and r
r
r = r
1
·· ·r
|r
r
r|
of a protein
and RNA, find whether or not p
i
and r
j
interacts for
all i and j. We briefly review the CRF-based method
and an evolutionary measurement MI
p
(mutual infor-
mation improved) proposed by (Dunn et al., 2008) be-
tween positions of amino acids and bases, and the lat-
ter is also used by our method. After that, we explain
our neural network architecture.
2.1 Evolutionary Measurement
One method of measuring coevolutionary relationship
between positions of a residue and a base in protein
and RNA sequences is to calculate mutual informa-
tion from multiple sequence alignments. MI
p
was
developed to improve protein residue-residue contact
prediction by subtracting a bias from mutual informa-
tion. Suppose that we have two multiple alignments
for protein and RNA sequences, p
p
p = p
1
·· · p
|p
p
p|
and
r
r
r = r
1
·· ·r
|r
r
r|
, respectively, where |p
p
p| means the length
of p
p
p. If i-th residue p
i
and j-th base r
j
interact with
each other to maintain a biological system in an indi-
vidual organism, there must be some relationship be-
tween them. Then, mutual information between po-
sitions i and j is defined by m
i j
=
∑
a∈A
∑
b∈B
Pr(p
i
=
a, r
j
= b) log
Pr(p
i
=a,r
j
=b)
Pr(p
i
=a)Pr(r
j
=b)
, where A and B denote
sets of amino acids and bases, respectively. MI
p
was
modified as m
i j
−
∑
|p
p
p|
i=1
m
i j
∑
|r
r
r|
j=1
m
i j
∑
|p
p
p|
i=1
∑
|r
r
r|
j=1
m
i j
for our purpose.
2.2 Conditional Random Field
(CRF)-based Method
Conditional random fields (CRFs) were proposed
by extending Markov random fields (Lafferty
et al., 2001). The CRF with a strictly positive
density in the previous study was defined by
Pr(x
i j
|x
x
x
N
i j
, m
m
m, p
p
p, r
r
r) =
1
Z
i j
exp
w
w
w
T
f
f
f
f
i j
(x
x
x, m
m
m, p
p
p, r
r
r) +
w
w
w
T
g
∑
(k,l)∈N
i j
g
g
g
i jkl
(x
x
x, m
m
m, p
p
p, r
r
r)
, where x
i j
= 1
means that i-th residue and j-th base in-
teract with each other, x
i j
= −1 otherwise,
f
f
f
i j
(x
x
x, m
m
m, p
p
p, r
r
r) = x
i j
1
m
i j
max
(k,l)∈N
i j
m
kl
min
(k,l)∈N
i j
m
kl
⊕ δ
(p
i
,r
j
)
,
g
g
g
i jkl
(x
x
x, m
m
m, p
p
p, r
r
r) = x
i j
x
kl
|m
i j
− m
kl
|
m
i j
m
kl
, δ
(p
i
,r
j
)
de-
notes a |A| × |B| dimensional vector which only the
element corresponding to the amino acid p
i
and base
r
j
is one, N
i j
for i = 1, ·· · , |p
p
p| and j = 1, ··· , |r
r
r| de-
notes a set of adjacent pairs of (i, j), and was defined
by (i ± 1, j), (i, j ± 1) (see Figure 1). In this method,
m
i
0
j
0
at the gray square in the figure was given
as one of input data for a residue-base pair (i, j),
that is, (i
0
, j
0
) ∈ {(i, j)} ∪ N
i j
∪
S
(k,l)∈N
i j
N
kl
. The
probability model contains dependency relationships
between positions (i, j) and (i
0
, j
0
) ∈ N
i j
.
Parameters w
w
w
f
and w
w
w
g
in the CRF model are deter-
mined by maximizing the pseudo-likelihood function,
L(θ) =
∏
n
∏
|p
p
p|
i=1
∏
|r
r
r|
j=1
Pr(x
(n)
i j
|x
x
x
(n)
N
i j
, m
m
m
(n)
, p
p
p
(n)
, r
r
r
(n)
, θ)
given x
x
x
(n)
, m
m
m
(n)
for each sequence pair p
p
p
(n)
, r
r
r
(n)
.
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
164

i
j
3URWHLQVHTXHQFH
51$VHTXHQFH
/
7
5
'
0
*
&
*
&
&
i
j
Figure 1: Illustration of adjacent pairs of (i, j). The posi-
tions (i
0
, j
0
) at the gray squares are included in N
i j
.
2.3 Neural Network Architecture
We propose a neural network with five layers as
shown in Figure 2. The input layer has (2k
m
+ 1)
2
+
(|A|+|B|)(2k
s
+1) neurons having evolutionary mea-
surement values of pairs (i
0
, j
0
) for all i − k
m
≤ i
0
≤
i+k
m
, j −k
m
≤ j
0
≤ j+k
m
, and one-hot vectors corre-
sponding to subsequences from p
i−k
s
to p
i+k
s
of pro-
tein p
p
p and from r
j−k
s
to r
j+k
s
of RNA r
r
r, where k
m
and k
s
are constant integers, and a one-hot vector for
a constant number c is defined as a vector that only
c-th element is one and the others are zero. For exam-
ple, when k
m
= 2, m
i
0
j
0
at the gray and white square
in Figure 1 is given with respect to (i, j) to the in-
put layer. In addition, amino acids can be classified,
and |A| can be equal or less than 20. The neural net-
work has three hidden layers with n
1
, n
2
, and n
3
neu-
rons, respectively. The output layer has two neurons
corresponding to the two classification results. All
successive layers are fully connected and the recti-
fied linear unit (ReLU) (Glorot et al., 2011) defined
by f (x) = max{0, x} is applied to the output of each
neuron except the final layer. The softmax function
is applied in the final layer. In addition, bias variables
are added to each neuron except the input layer. Then,
the total number of parameters is ((2k
m
+1)
2
+(|A| +
|B|)(2k
s
+1))n
1
+n
1
+
∑
3
i=1
{n
i
n
i+1
+n
i+1
}+2n
3
+2.
input layer hidden layers output layer
Figure 2: Illustration of the neural network with five layers.
3 RESULTS
For evaluation of the proposed method, we used the
same dataset as that in the previous study, which con-
sists of residue-base pairs included in thirteen protein-
RNA pairs in four complexes identified by ’1yl4’,
’2hgu’,’3kc4’, and ’3kcr’ in PDB (Rose et al., 2017)
as shown in Table 1.
Each line in the table shows a protein-RNA pair,
the PDB identifier, the chain identifier, the protein
and RNA sequences p
p
p, r
r
r in UniProt and GenBank
databases (The UniProt Consortium, 2017; Benson
et al., 2011), the length, and the number of contacts
between residues and bases in the protein and the
RNA. It was assumed that i-th residue and j-th base
interact with each other if the Euclidean distance be-
tween atoms of the residue and base is less than or
equal to 3
˚
A because the distances of hydrogen bonds
are about 2.7 to 2.9
˚
A.
We used multiple sequence alignments of Pfam
and Rfam databases (Finn et al., 2016; Kalvari et al.,
2018) for the protein p
p
p and RNA r
r
r to calculate MI
p
.
We examined classifications of amino acids with 8,
10, and 15 groups according to the study (Murphy
et al., 2000), that is, |A| took 8, 10, 15, and 20, and
MI
p
was calculated based on each case of A (see Ta-
ble 2).
We performed cross-validation procedures, and
took the average of AUC scores as well as the previ-
ous study, where among thirteen protein-RNA pairs in
Table 1, all residue-base pairs included in one protein-
RNA pair were used for test, and the others were used
for training. We examined the window size by varying
the values of k
m
and k
s
, and set n
1
= 750, n
2
= 680,
and n
3
= 250. We used the tensorflow-gpu library
(version 1.4.1) to minimize the cross entropy of the
output and to calculate the AUC score (Abadi et al.,
2015).
Table 3 shows the results on average AUC
scores by the CRF-based method and the proposed
fully-connected neural network method in the cases
(k
m
, k
s
) = (1, 2), (2, 1), (2, 2). In the three cases, the
AUC scores in the case of (2, 1) were better than those
in other cases in the same classification of amino
acids. It implies that amino acids and bases at po-
sitions (i ± 2, j ± 2) far from the position of inter-
est (i, j) were not effective to enhance the predic-
tion accuracy. On the other hand, the evolutionary
measurement MI
p
between positions (i ± 2, j
0
) and
(i
0
, j ±2) for i− 2 ≤ i
0
≤ i +2, j −2 ≤ j
0
≤ j + 2 were
useful. It is expected that the neural network with
(k
m
, k
s
) = (2, 2) would be equivalent to the neural net-
work with (2, 1) if appropriate parameters are esti-
mated as zero, and the AUC score with (2, 2) would
Artificial Neural Network Approach to Prediction of Protein-RNA Residue-base Contacts
165
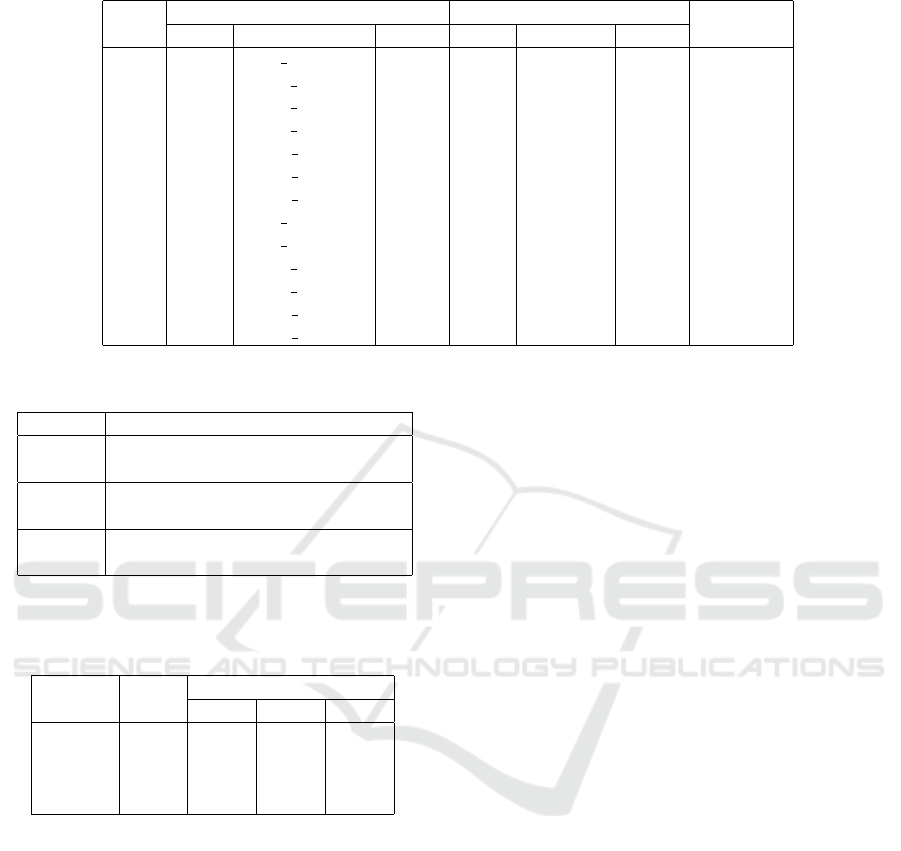
Table 1: Dataset of the residue-base pairs in protein-RNA complexes.
PDB Protein sequence RNA sequence # contacts
chain UniProt length chain GenBank length
1yl4 K RS8 THET8 135 A M26923 1889 29
1yl4 M RS10 THET8 97 A M26923 1711 20
1yl4 O RS12 THET8 122 A M26923 1972 45
1yl4 T RS17 THET8 69 A M26923 1690 29
2hgu R RL18 THETH 110 B X01554 1543 28
2hgu Z RL27 THET8 81 A X12612 1356 20
2hgu 5 RL33 THET8 48 A X12612 1445 18
3kc4 E RS5 ECOLI 67 A J01695 1701 13
3kc4 G RS7 ECOLI 147 A J01695 1941 25
3kc4 O RS15 ECO57 83 A J01695 1821 21
3kc4 Q RS17 ECOLI 69 A J01695 1690 18
3kcr W RL27 ECOLI 77 8 J01695 1356 18
3kcr 3 RL35 ECOLI 61 8 J01695 1337 12
Table 2: Grouping of amino acids (Murphy et al., 2000).
#groups groups of amino acids
8 (MLVIC) (GA) (TS) (P) (FYW)
(DENQ) (RK) (H)
10 (MLVI) (C) (G) (A) (TS) (P)
(FYW) (DENQ) (RK) (H)
15 (MLVI) (C) (G) (A) (T) (S) (P)
(FY) (W) (D) (E) (N) (Q) (RK) (H)
Table 3: Results on average AUC scores by the CRF-
based method and the proposed neural network method with
(k
m
, k
s
) = (1, 2), (2, 1), (2, 2).
#groups CRF Proposed
(1,2) (2,1) (2,2)
8 0.692 0.671 0.674 0.668
10 0.699 0.673 0.684 0.681
15 0.699 0.670 0.711 0.682
20 0.693 0.665 0.690 0.678
be larger than or equal to that with (2, 1). However,
the AUC score with (2, 1) was larger than that with
(2, 2).
For classification of amino acids, as reported in
the previous study, the AUC score with 15 groups
was better than others in almost all cases. In addition,
the AUC score by our method with (k
m
, k
s
) = (2, 1)
and 15 groups was better than that by the CRF-based
method.
4 CONCLUSIONS
We proposed a neural network approach to prediction
of protein-RNA residue-base contacts. In the neu-
ral network, neurons between successive layers were
fully connected, and the ReLU activation function
was applied. From the cross-validation computational
experiments to evaluate the proposed method, the re-
sults show that in terms of the area under the receiver
operating characteristic curve (AUC), the predictive
performance of our proposed method was compara-
ble or better than those of the CRF-based method.
As future work, other types of advanced neural net-
works should be examined for further improvement
of prediction accuracy, and for understanding inter-
actions between residues and bases in detail. In
our method, subsequences as long as motifs cannot
be dealt. Therefore, we would like to improve our
method to deal with longer subsequences.
ACKNOWLEDGEMENTS
This work was partially supported by Grants-in-Aid
#16K00392 and #16KT0020 from JSPS, Japan.
REFERENCES
Abadi, M., Agarwal, A., Barham, P., Brevdo, E., Chen, Z.,
Citro, C., Corrado, G. S., Davis, A., Dean, J., Devin,
M., Ghemawat, S., Goodfellow, I., Harp, A., Irving,
G., Isard, M., Jia, Y., Jozefowicz, R., Kaiser, L., Kud-
lur, M., Levenberg, J., Man
´
e, D., Monga, R., Moore,
S., Murray, D., Olah, C., Schuster, M., Shlens, J.,
Steiner, B., Sutskever, I., Talwar, K., Tucker, P., Van-
houcke, V., Vasudevan, V., Vi
´
egas, F., Vinyals, O.,
Warden, P., Wattenberg, M., Wicke, M., Yu, Y., and
Zheng, X. (2015). TensorFlow: Large-scale machine
learning on heterogeneous systems. Software avail-
able from tensorflow.org.
Alipanahi, B., Delong, A., Weirauch, M., and Frey, B.
(2015). Predicting the sequence specificities of DNA-
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
166

and RNA-binding proteins by deep learning. Nature
Biotechnology, 33:831–838.
Benson, D., Karsch-Mizrachi, I., Lipman, D., Ostell, J., and
Sayers, E. (2011). Genbank. Nucleic Acids Research,
39:D32–D37.
Beyer, A., Christensen, M., Walker, B., and LeStourgeon,
W. (1977). Identification and characterization of the
packaging proteins of core 40S hnRNP particles. Cell,
11:127–138.
Breiman, L. (2001). Random forests. Machine Learning,
45:5–32.
Dunn, S., Wahl, L., and Gloor, G. (2008). Mutual in-
formation without the influence of phylogeny or en-
tropy dramatically improves residue contact predic-
tion. Bioinformatics, 24:333–340.
Feng, G.-S., Chong, K., Kumar, A., and Williams, B.
(1992). Identification of double-stranded RNA-
binding domains in the interferon-induced double-
stranded RNA-activated p68 kinase. Proc. Natl. Acad.
Sci. USA, 89:5447–5451.
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R.,
Mistry, J., Mitchell, A. L., Potter, S. C., Punta, M.,
Qureshi, M., Sangrador-Vegas, A., Salazar, G. A.,
Tate, J., and Bateman, A. (2016). The Pfam protein
families database: towards a more sustainable future.
Nucleic Acids Research, 44(D1):D279–D285.
Glisovic, T., Bachorik, J., Yong, J., and Dreyfuss, G. (2008).
RNA-binding proteins and post-transcriptional gene
regulation. FEBS Letters, 582:1977–1986.
Glorot, X., Bordes, A., and Bengio, Y. (2011). Deep sparse
rectifier neural networks. In 14th International Con-
ference on Artificial Intelligence and Statistics, pages
315–323.
Gupta, A. and Gribskov, M. (2011). The role of RNA
sequence and structure in RNA-protein interactions.
Journal of Molecular Biology, 409:574–587.
Hall, T. (2005). Multiple modes of RNA recognition by
zinc finger proteins. Current Opinion in Structural
Biology, 15:367–373.
Hayashida, M., Kamada, M., Song, J., and Akutsu, T.
(2013). Prediction of protein-RNA residue-base con-
tacts using two-dimensional conditional random field
with the lasso. BMC Systems Biology, 7(Suppl 2):S15.
Hayashida, M., Okada, N., Kamada, M., and Koyano, H.
(2018). Improving conditional random field model
for prediction of protein-RNA residue-base contacts.
Quantitative Biology, 6:155–162.
Kalvari, I., Argasinska, J., Quinones-Olvera, N., Nawrocki,
E. P., Rivas, E., Eddy, S. R., Bateman, A., Finn, R. D.,
and Petrov, A. I. (2018). Rfam 13.0: shifting to a
genome-centric resource for non-coding RNA fami-
lies. Nucleic Acids Research, 46(D1):D335–D342.
Kedersha, N., Gupta, M., Li, W., Miller, I., and Anderson,
P. (1999). RNA-binding proteins TIA-1 and TIAR
link the phosphorylation of eIF-2α to the assembly of
mammalian stress granules. Journal of Cell Biology,
147:1431–1441.
Lafferty, J., McCallum, A., and Pereira, F. (2001). Con-
ditional random fields: Probabilistic models for seg-
menting and labeling sequence data. In Proc. Int.
Conf. on Machine Learning.
LeCun, Y., Bengio, Y., and Hinton, G. (2015). Deep learn-
ing. Nature, 521:436–444.
Murphy, L., Wallqvist, A., and Levy, R. (2000). Simpli-
fied amino acid alphabets for protein fold recognition
and implications for folding. Protein Engineering,
13:149–152.
Rose, P. W., Prli
´
c, A., Altunkaya, A., Bi, C., Bradley, A. R.,
Christie, C. H., Costanzo, L. D., Duarte, J. M., Dutta,
S., Feng, Z., Green, R. K., Goodsell, D. S., Hud-
son, B., Kalro, T., Lowe, R., Peisach, E., Randle,
C., Rose, A. S., Shao, C., Tao, Y.-P., Valasatava, Y.,
Voigt, M., Westbrook, J. D., Woo, J., Yang, H., Young,
J. Y., Zardecki, C., Berman, H. M., and Burley, S. K.
(2017). The RCSB protein data bank: integrative view
of protein, gene and 3D structural information. Nu-
cleic Acids Research, 45(D1):D271–D281.
Sharan, M., F
¨
orstner, K., Eulalio, A., and Vogel, J. (2017).
APRICOT: an integrated computational pipeline for
the sequence-based identification and characterization
of RNA-binding proteins. Nucleic Acids Research,
45:e96.
Siomi, H., Matunis, M., Michael, W., and Dreyfuss, G.
(1993). The pre-mRNA binding K protein contains
a novel evolutionary conserved motif. Nucleic Acids
Research, 21:1193–1198.
Sun, M., Wang, X., Zou, C., He, Z., Liu, W., and Li, H.
(2016). Accurate prediction of RNA-binding protein
residues with two discriminative structural descrip-
tors. BMC Bioinformatics, 17(1):231.
Tang, Y., Liu, D., Wang, Z., Wen, T., and Deng, L. (2017).
A boosting approach for prediction of protein-RNA
binding residues. BMC Bioinformatics, 18(13):465.
The UniProt Consortium (2017). UniProt: the univer-
sal protein knowledgebase. Nucleic Acids Research,
45:D158–D169.
Weirauch, M. T., Cote, A., Norel, R., Annala, M., Zhao,
Y., Riley, T. R., Saez-Rodriguez, J., Cokelaer, T., Ve-
denko, A., Talukder, S., Consortium, D., Bussemaker,
H. J., Morris, Q. D., Bulyk, M. L., Stolovitzky, G., and
Hughes, T. R. (2013). Evaluation of methods for mod-
eling transcription factor sequence specificity. Nature
Biotechnology, 31:126–134.
Zeng, H., Edwards, M., Liu, G., and Gifford, D.
(2016). Convolutional neural network architectures
for predicting DNA-protein binding. Bioinformatics,
32:i121–i127.
Zhao, Y., Stormo, G., Feature, N., and Eisenstein, M.
(2011). Quantitative analysis demonstrates most tran-
scription factors require only simple models of speci-
ficity. Nature Biotechnology, 29:480–483.
Artificial Neural Network Approach to Prediction of Protein-RNA Residue-base Contacts
167
