
Indexing k-mers in Linear-space for Quality Value Compression
Yoshihiro Shibuya
1,2
and Matteo Comin
1
1
Department of Information Engineering, University of Padua, via Gradenigo 6B, Padua, Italy
2
Laboratoire d’Informatique Gaspard-Monge (LIGM), University Paris-Est Marne-la-Vall´ee,
Bˆatiment Copernic - 5, bd Descartes, Champs sur Marne, France
Keywords:
k-mers, Indexing, Quality Score, Read Compression.
Abstract:
Many bioinformatics tools heavily rely on k-mer dictionaries to describe the composition of sequences and
allow for faster reference-free algorithms or look-ups. Unfortunately, naive k-mer dictionaries are very mem-
ory inefficient, requiring very large amount of storage space to save each k-mer. This problem is generally
worsened by the necessity of an index for fast queries. In this work we discuss how to build an indexed linear
reference containing a set of input k-mers, and its application to the compression of quality score in FASTQ
files. Most of the entropy of sequencing data lies in the quality scores, and thus they are difficult to compress.
Here, we present an application to improve the compressibility of quality values while preserving the infor-
mation for SNPs calling. We show how a dictionary of significant k-mers, obtained from SNPs databases and
multiple genomes, can be indexed in linear space and used to improve the compression of quality value.
Availability: the software is freely available at https://github.com/yhhshb/yalff.
1 INTRODUCTION
The compression of DNA is usually as simple as as-
signing a two bit encoding to each of the four bases.
This encoding achieve almost similar results to stan-
dard lossless compressors (Malysa et al., 2015). On
the other hand, the quality values, produced by se-
quencing technologies, span a wider range of values,
and when compressed they can sum up to about 70%
of the total space to encode a FASTQ file (Greenfield
et al., 2016). Quality valuesare usually encoded using
the Phred system (Ewing et al., 1998). Quality val-
ues are often essential for assessing sequence quality,
mapping reads to a reference genome, detecting muta-
tions for genotyping, assembling genomic sequences,
reads clustering (Comin et al., 2014; Comin et al.,
2015) and comparison (Schimd and Comin, 2016).
Quality scores are more difficult to compress due
to a larger alphabet (63-94 in original form) and in-
trinsically have a higher entropy (Yu et al., 2015).
With lossless compression algorithms and entropy en-
coders reaching their theoretical limits and delivering
only moderate compression ratios (Bonfield and Ma-
honey, 2013), there is a growing interest to develop
lossy compression schemes to improve compressibil-
ity further.
To further reduce the file sizes, Illumina proposed
a binning method to reduce the number of differ-
ent quality values from 42 to 8 (Illumina8bin, 2011).
With this proposal, Illumina opened the doors for al-
lowing lossy compression of the quality values. An-
other approach called P-Block (C´anovas et al., 2014)
involves local quantization so that a representative
quality score replaces a contiguous set of quality
scores that are within a fixed distance of the represen-
tative score. Similarly, the R-Block (C´anovas et al.,
2014) scheme replaces contiguous quality scores that
are within a fixed relative distance of a representa-
tive score. Other lossy approaches improve com-
pressibility and preserve higher fidelity by minimiz-
ing a distortion metric such as mean-squared-error
or L1-based errors (Qualcomp and QVZ) ((Malysa
et al., 2015) (Ochoa et al., 2013)). The drawback
of lossy compression of quality values is that down-
stream analysis could be affected by the loss in-
curred with this type of compression. This could
be the case for the above methods that process only
the string of quality scores, without considering the
DNA sequence associated to the read. However, (Yu
et al., 2015), (Ochoa et al., 2017) and (Greenfield
et al., 2016) showed that quality values compressed
with more advanced methods could achieve not only
a better performance in downstream analyses than
Illumina-binnedquality values, but even better perfor-
mance than the original quality values in some cases
because these methods remove noise from the data.
Shibuya, Y. and Comin, M.
Indexing k-mers in Linear-space for Quality Value Compression.
DOI: 10.5220/0007369100210029
In Proceedings of the 12th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2019), pages 21-29
ISBN: 978-989-758-353-7
Copyright
c
2019 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
21

The most promising methods are those using both
sequence and quality information. Leon (Benoit et al.,
2015) constructs a reference from the input reads in
the form of a bloom filter compressed de-Bruijn graph
and then maps each nucleotide sequence as a path in
the graph. If a base is covered by a sufficiently large
number of k-mers from the reference its quality is
set at a fixed high value. Among the most interest-
ing tools, Quartz (Yu et al., 2015), similarly to Leon,
relies on an external reference to decide if a given
nucleotide is wrong or not. This reference database
is implemented as list of 2497777428 k-mers stored
explicitly, that requires 24GB when gzipped. Simi-
larly, GeneCodeq (Greenfield et al., 2016) also has
a list of k-mers as ground truth, but the algorithm
involved during quality compression, i.e. smooth-
ing, is more complex than Quartz. Each base has
its associated error probability recalculated using a
Bayesian framework and the smoothing takes place
only if the new quality is greater than the old one.
Both Quartz (Yu et al., 2015) and GeneCodeq (Green-
field et al., 2016) require a machine with at least
32GB of RAM, because of the size of the reference
database. In (Shibuya and Comin, 2018) we have pro-
posed a method for quality value compression based
on a single reference genome. However, the authors
of (Yu et al., 2015) demonstrated that the use of sig-
nificant k-mers coming from multiple genomes or
SNPs databases, are beneficial for quality score com-
pression and SNPs calling. Hence, in this paper we
explore the use of multiple sources of mutations, i.e.
SNP databases, for quality value compression.
The most common procedure to obtain a reference
list of k-mers from a set of sequences is by a k-mer
counting procedure. Most of the existing implemen-
tations rely on some hashing scheme. The hashing
usually involves a simple application of a hash func-
tion to each k-mer to obtain a numeric value which
is then used to access a hash table where the actual
counters are kept. There exist efficient implementa-
tions of hashing functions which focus on optimizing
and accelerating the generation of the hash value by
using the previous computed hash like in (Mohamadi
et al., 2016) or (Girotto et al., 2018b; Girotto et al.,
2018a). On the other hand, a very fast implementation
of a hash table can be found in Jellyfish (Marc¸ais and
Kingsford, 2011) which uses a lock-free table allow-
ing access from multiple threads if they do not collide
in the same bucket for writing operations. Another ap-
proach is found in (Rizk et al., 2013) and involves the
use of minimizers, a type of Locality Sensitive Hash-
ing to efficiently group similar k-mers into buckets.
The next step usually involves some sort of sort-
ing and indexing for allowing fast retrieval of some
particular k-mer (or its neighbors) and link this infor-
mation to the position in the original sequence. This
step is necessary whenever the counting procedure is
the first step of a more complex pipeline. It is not so
uncommon for some applications to index a sequence
using the positions of each of its k-mers (Greenfield
et al., 2016; Shajii et al., 2016). While this approach
is advantageous when a proper counting of the k-mers
is required it is not necessary in general when the final
goal is to support set queries and/or retrieval of a spe-
cific sequence or its position inside the original string.
For this purpose, it is generally possible to strip a dic-
tionary of k-mers of the counters to obtain a smaller
representation. Unfortunately, even in the hypothe-
sis of removing all the under-represented k-mers and
all counters, the space required by a full table is still
high. For example, LAVA (Shajii et al., 2016) uses
a databases of k-mers extracted from dbSNP (Sherry
et al., 2001), because of the large size of this database
it requires 60GB of ram. In fact, for a DNA sequence
of length n the potential number of k-mers is n−k+ 1
each of length k for a total amount of space in the or-
der of O(k(n− k + 1)) = O(kn) bytes.
If the k-mer table has to be transmitted a naive
solution would be to compress it using a standard
compressor such as
gzip
or
xz
. This procedure only
works for moving the database from one place to an-
other because to fully restore its functionality it must
be decompressed anyway. Another question is how
efficient the standard compressors (or an two bit en-
coding) are in reducing the size of the table. What
is the best way to compress a set of k-mers? Is
it possible to use them for an optimal compression
scheme removing redundancy and exploiting the pe-
culiar structure of each k-mer and its relation with the
others?
To answer this question in this work we analyze a
particular application of k-mer dictionaries, that is the
compression of quality scores. In this work we inves-
tigate how to construct a linear string, or set of strings,
that contains all input k-mers. In particular we will
consider a special set of k-mers extracted from pop-
ular SNPs databases like dbSNP (Sherry et al., 2001)
or Affymetrix Genome-Wide Human SNP Array 6.0.
In the following sections we present an application of
a reassembly procedure to reduce a dictionary of k-
mers into a set of sequences guaranteed to contain all
the k-mers. This procedure will allow us to store the
whole dictionary in linear form reducing the mem-
ory requirements from O(kn) to O(n) without losing
information. For querying the resulting dictionary we
will index it with the FM-Index, and use the FM-index
for quality value compression.
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
22

2 METHODS
The main insights in order to reduce the size of the
dictionary is that most of the information carried by a
k-mer stored explicitly is redundant. This intuition is
easily explained by recalling the k-mer counting pro-
cedure itself. All the k-mers counted comes from a
set sequences and the counting procedure is only nec-
essary to remove the wrong ones. There is no need
to keep the k-mers explicitly stored to answer simple
yes/no queries over their set. Given two consecutive
k-mers it is possible to reassemble them into a single
(k+1)-mer thus reducing the storage requirement by
k-1 bases.
2.1 Dictionary Reassembly
There exists two methods to build a dictionary string
comprising all the k-mers needed for quality smooth-
ing. The first is a k-mer counting procedure followed
by an assembly (Figure 1 top left). If the counting is
performed on a set of real datasets of reads then a fil-
tering of the k-mers whose counter is below a certain
threshold might be required to prevent false positives
to be used as ground truth, as in (Yu et al., 2015). The
filter is effective only if the datasets used have a very
high coverage, leading to increased memory and time
requirements for this step.
The reassembly step can be carried out on almost
all dictionaries regardless of the tools used to gener-
ate them, leading to a linear sequence, or set of se-
quences, that contains all the input k-mers. This re-
quirement is what makes this step different from a
common de-novo assembly where the k-mers that do
not align well with the graph are discarded leading
to k-mers not present in the final reconstructed se-
quence. In general, this procedure will output more
than one sequence because of the uniqueness of each
k-mer in the resulting strings. Some redundantk-mers
could allow for the concatenation of multiple contigs
into one. The strings obtained are generally stored
into a simple FASTA file where the identifiers can
lead to unwanted memory usage especially if there
are a very large number of short contigs. The mem-
ory requirements can worsening during the indexing
of the FASTA file by adding information to account
for the boundaries of each sequence. Simply con-
catenating each conting to each other produce k-1
spurious k-mers for each junction which might be a
source of potential false-positives during the applica-
tion of quality value compression. The software used
to perform this step is the greedy ProphAsm assem-
bler (ProphAsm) used in the Prophyle metagenomic
suite (Bˇrinda, 2016; Bˇrinda et al., 2017). In contrast
to the similar software BCalm (Chikhi et al., 2016) it
doesn’t stop at each discordant new k-mer producing
less and longer contigs.
If the previous method does not lead to a signifi-
cant reduction of space compared to an already avail-
able reference genome (Figure 1 bottom left) then
the latter one can be the right choice. The previ-
ous method of construction will probably lead to a
set of strings very similar to the reference genome of
the organism even when applied in an optimal setting
(when the k-mers can be aligned into few contigs).
It is true that a counting procedure involving many
sequences will include many variants in the final dic-
tionary string but nonetheless it is very convenient to
use a reference genome directly.
Both method can be extended for accounting for
known lists of SNPs. The first one can include ad-
ditional k-mers during the counting procedure and
reassembly the whole dictionary back into a string.
These additional k-mers can be extracted from a list
of known SNPs, like in LAVA (Shajii et al., 2016),
where for each SNP we can we take the k-mers that
overlap with the SNP with the reference allele re-
placed by the alternate. If the starting point is an al-
ready assembled set of dictionary strings it is possible
to simply add the relevant k-mers at the end of one
contig by concatenation or by maintaining the list of
known SNPs as is and load it together with the refer-
ence string. This work uses the first method because
as described in the Results section the realigned dic-
tionary contains significant k-mers coming from mul-
tiple human genomes, extracted from dbSNP (Sherry
et al., 2001) and Affymetrix Genome-Wide Human
SNP Array 6.0.
The problem of indexing a set of reference se-
quences in minute space, while providing full search
capability, has been widely studied and efficient data
structure are now available. The data structure cho-
sen for this purpose is the FM-Index (Ferragina and
Manzini, 2000; Ferragina and Manzini, 2005) which
is based on the Burrows-Wheeler transform (BWT)
(Burrows and Wheeler, 1994) of a sequence. The
FM-index, and its variants, are now at the basis of
many algorithms in the field of sequence analysis. For
example, one of the most used tool for reads map-
ping, BWA (Li and Durbin, 2010), is based on the
FM-index and it requires as input the FM-index of the
reference genome. For its simplicity, its relative easy
installation and its widespread usage, we decided to
use the BWA for indexing and querying our list of
sequences. The FM-index will be used to search for
k-mers. The procedure to retrieve the position of a
k-mer is the enhanced backward search algorithm de-
scribed in (Li and Durbin, 2009), that is also able to
Indexing k-mers in Linear-space for Quality Value Compression
23

Figure 1: Examples of dictionary string construction. The first method employs a k-mer counting procedure, the second one
uses an already available reference genome and other reference sequences. Additional information, e.g. regarding SNPs data,
can be included in the dictionary string by concatenation. In the last step the dictionary string is indexed using an FM-Index.
Figure 2: An example of quality smoothing by YALFF including both mismatches with the k-mers DB and low quality values.
account for mismatches. In our case we will search if
a k-mer is present in the reference genome with up to
one mismatch.
In our work for compressing quality scores
(Shibuya and Comin, 2018) the use of BWA also had
the collateral advantage of not requiring a separate
indexed FASTA for compression instead sharing the
same indexed reference genome for reads alignments.
In this work the indexed sequences are not equivalent
to a reference genome, but instead they represent a
set of k-mers that uniquely identifies known variants
from dbSNP and Affymetrix SNPs databases.
2.2 Quality Value Compression based
on k-mers
We evaluate the quality of the k-mers dictionary for
the compression of quality values. This work extends
our previous work on YALFF (Shibuya and Comin,
2018) where we presented a novel algorithm for com-
pressing quality strings without losing precision dur-
ing SNPs calling. The main differences are in the ref-
erence sequence used for compression. In (Shibuya
and Comin, 2018) we compress a FASTQ file using
always a single reference genome (i.g. HG38), while
in this study we build a sequence dictionary based
on a set of significant k-mers that appear in multiple
genomes coming from the 1000 Genome Project, the
dbSNP, and the Affymetrix SNPs databases. To keep
the paper self-contained here we briefly describe the
YALFF algorithm.
The method YALFF employs a dictionary of k-
mers to assess if a base of a read is correct. A reads is
decomposed into its constituent k-mers and they are
searched in the reference dictionary. Given a base of
the read, if all k-mers that cover the base are found
in the dictionary then we assume that the base is cor-
rect and thus we can modify (smooth) the correspond-
ing quality score to a fixed high value, see Figure
2 for an example. In the first version of YALFF,
the database against which the k-mers are checked
is the well known human reference genome
hg38
in
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
24

the form of a BWA index. A summary of the algo-
rithm is illustrated in Figure 3. The threshold should
be chosen depending on if it is necessary to avoid as
much distortion as possible or if compression is con-
sidered much more important. As a rule of thumb
a higher threshold maintains more quality values un-
changed but this leads to an increase in the entropy of
the output file. A good value found for this study was
a quality value equal to the character ’ (apex) which
corresponds to a probability of error of 0.25119.
Quartz (Yu et al., 2015) is another FASTQ com-
pressor which explicitly stores all the k-mers in a
hash table. Quartz requires 25GB of RAM whereas
YALFF only need 5.7GB. The k-mer counting pro-
cedure used to build the default database of Quartz
was performed on multiple NGS reads coming from
different individuals of the 1000 Genome Project and
therefore contains more significant k-mers compared
to the simple reference genome. Similarly in LAVA
(Shajii et al., 2016), the authors use two popular
SNPs databases like dbSNP (Sherry et al., 2001) and
Affymetrix Genome-Wide Human SNP Array to se-
lect a set of k-mers that uniquely identify these muta-
tions. Here, we will use these set of k-mers instead of
a single reference genome.
All subsequent results use k equal to 32, the length
used by other studies (Yu et al., 2015; Greenfield
et al., 2016; Shibuya and Comin, 2018). 32 has been
chosen because the k-mers should be long enough
to ensure that the number of all possible k-mers is
much larger than the number of unique k-mers in the
genome, so as to ensure incidental collisions between
unrelated k-mers are rare. Also, k-mer length should
ideally be a multiple of four, since a 4bp length DNA
sequence can be represented by a single byte and dis-
criminating between forward and reverse strand is not
important in this application (it is only required a
yes/no response to a query). A 32-mer satisfies these
constraints (Yu et al., 2015; Greenfield et al., 2016);
it is represented by a single 64-bit integer, with a rel-
atively low probability of containing more than one
sequencing error with Illumina sequences, as well as
resulting in few k-mer collisions.
2.3 Datasets, Pipeline and Parameters
The dataset used in this study is a set of real reads
(NA12878) from the 1000 Genomes Project (Con-
sortium, 2012). Only the two paired end archives
were used (namely SRR622461
1.filt.fastq.gz and
SRR622561
2.filt.fast.gz) for the evaluation, while
the third containing unpaired reads were discarded.
For validation, we used an up-to-date high-quality
genotype annotation generated by the Genome in a
Bottle Consortium (Zook et al., 2014). The GIAB
gold standard contains validated genotype informa-
tion for NA12878, from 14 sequencing datasets with
five sequencing technologies, seven read mappers and
three different variant callers. To measure accuracy,
we use loci in the SNP list which are also genotyped
in the GIAB gold standard (so called high confident
regions) (SNPs, 2018). This dataset has been widely
used for benchmarking in other papers (Yu et al.,
2015; Shajii et al., 2016),
One of the dictionary string used as a reference
is the human genome reference FASTA file
hg38.fa
downloaded from (HG38, 2018). The other k-mers
databases are generated from dbSNPs (Sherry et al.,
2001) and Affymetrix Genome-Wide Human SNP
Array 6.0. Although YALFF can be run on a normal
laptop the ProphAsm assembler requires a powerful
computer to load all the k-mers it needs to reassem-
bly. For this reason all tests were performed on a
14 lame blade cluster DELL PowerEdge M600 where
each lame is equipped with two Intel Xeon E5450 at
3.00 GHz, 16GB RAM and two 72GB hard disk in
RAID-1 (mirroring).
The basic idea is to compare the compression re-
sults for the given dataset using both the original
reference genome and the re-assembled dictionary
of k-mers of dbSNPs and Affymetrix Genome-Wide
Human SNP Array with YALFF’s algorithm. The
performance evaluation of the smoothing procedure
alone compares the number of retrieved SNPs from
a smoothed FASTQ to the ground truth associated to
the original dataset, with each set of variants (stored
in the output VCF file) compared against the consen-
sus set of variants. Each evaluation comprises three
metrics: Precision, Recall and F-Measure. Once these
parameters have been computed for using both dictio-
naries they can be compared each other to assess how
the additional variants inside the k-mers database can
influence a smoothing algorithm and the linear repre-
sentation of the k-mers set. The genotyping pipeline
is implemented as a single bash script which uses
bwa
mem
for alignments,
bcftools
for SNP calling and
rtgplot
for evaluation.
The exact commands are reported for each pro-
gram used outside the evaluation pipeline:
• ./print quartz dict dec200.bin.sorted | prophasm -
k 32 -i - -o <Output FASTA>
• cat <Input file> | ./YALFF -d hg38.fa -g \’ -t 1 >
<Output file>
Where
print quartz dict
is a simple utility
written in C that prints each k-mer of Quartz’s binary
dictionary to the standard output.
Indexing k-mers in Linear-space for Quality Value Compression
25
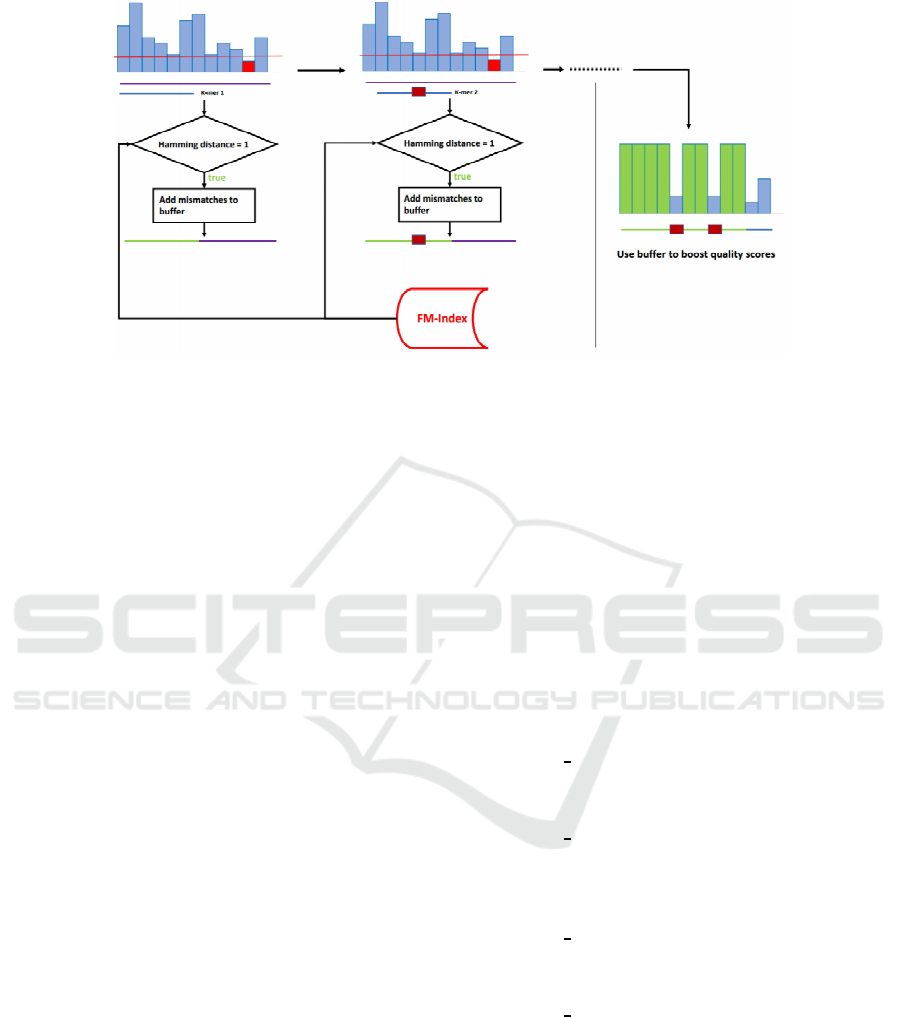
Figure 3: Diagram of YALFF’s inner workings.
3 RESULTS
In this section the effects of the different k-mers
databases are compared in terms of genotyping accu-
racy and compression of the database itself and the
input FASTQ files. In order to give some context
to the measurements related to the unprocessed input
and Quartz are also reported from the YALFF paper.
3.1 Effects on Genotyping Accuracy
The performance evaluation of the algorithms com-
pares the number of retrieved SNPs from a smoothed
file to the ground truth. The latter comes with the
original dataset and contains a set of variants val-
idated using the genetic information of the parents
whose child is the individual providing the source of
the dataset. Each set of variants is stored in a VCF file
analyzable with the software described before.
The benchmarking tools output the following val-
ues:
• True Positives (T.P.): All those variants that are
both in the consensus set and in the set of called
variants.
• False Positives (F.P.): All those variants that are in
the called set of variants but not in the consensus
set.
• False Negatives (F.N.): All those variants that are
in the consensus set but not in the set of called
variants.
These values are used to compute the following
three metrics:
• Recall: This is the proportion of called variants
that are included in the consensus set; that is, R =
T.P./(T.P. + F.N.),
• Precision: This is the proportion of consensus
variants that are called by the variant calling
pipeline; that is, P = T.P./(T.P. + F.P.).
• F-Measure: The harmonic mean of precision and
recall; that is, F − Measure = 2∗ (P∗ R)/(P+ R)
The following abbreviations are used in each ta-
ble:
• None: The original FASTQ files without any fur-
ther modification.
• Quartz: The result of smoothing the original input
dataset with Quartz and its standard k-mers dic-
tionary.
• YALFF
hg38: The result of smoothing the origi-
nal input dataset with YALFF and the hg38 refer-
ence genome as a database of k-mers.
• YALFF Affy: YALFF when using the re-
assembled database of significant k-mers from
Affymetrix Genome-Wide Human SNP Array
6.0.
• YALFF dbSNPs: YALFF when using the re-
assembled database of significant k-mers from db-
SNPs.
• YALFF All: YALFF when using the reassem-
bled database of k-mers from hg38, dbSNPs
and Affymetrix Genome-Wide Human SNP Ar-
ray 6.0.
In Table 1 are reported the results on the genotyp-
ing accuracy for the various tools.
Table 1 illustrates that Quartz is the most aggres-
sive in terms of raw numbers. Compared to the origi-
nal dataset it tends to add a very large number of True
Positives and False Positives while greatly decreas-
ing the False Negatives. This overall behavior would
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
26

Table 1: Comparison of the number of True Positives, False
Positives and False Negatives found by the SNP calling
pipeline after smoothing the input reads with the various
methods.
Method T.P. F.P. F.N.
None 2588159 219803 1493731
Quartz 2661218 237820 1420672
YALFF hg38 2603620 221368 1478264
YALFF Affy 2610302 222017 1471583
YALFF dbSNP 2645734 222820 1436155
YALFF All 2652457 224616 1429433
be more than adequate if well calibrated, but unfortu-
nately it tends to degrade the Precision. On the other
hand, the algorithm of YALFF
hg38 is more conser-
vative with a better precision and a lower number of
false positives, preferring to maintain as much as fi-
delity to the original as possible (see, Table 2).
We can observe that although YALFF
hg38
has a smaller F-Measure compared to Quartz, the
use of more informative k-mers dictionaries, e.g.
YALFF
dbSNP and YALFF All, increases the F-
measure to comparable values. In particular, these
two k-mers dictionaries appear to be only one that can
improve the precision with respect to the unprocessed
input. In summary, we can preserve an higher preci-
sion and a smaller number of false positives on SNP
calling. This is very important in many medical ap-
plications where false positives should be avoided.
Table 2: Equivalent representation of Table 1 in terms of
Precision, Recall and F-Measure.
Method Precision Recall F-Measure
None 0.9217 0.6341 0.7513
Quartz 0.9180 0.6520 0.7624
YALFF hg38 0.9216 0.6378 0.7539
YALFF Affy 0.9216 0.6395 0.7551
YALFF dbSNP 0.9223 0.6482 0.7613
YALFF All 0.9219 0.6498 0.7623
The reassembled dictionary of significant k-mers
allows YALFF to inherit some characteristics of its
rival. This can be easily noticed by looking at the
increased recall. The larger is the k-mers database,
the betters is the recall, reching its maximum with
YALFF
All. Unfortunately, the precision does not
follow a similar behavior, where YALFF
dbSNP is
the best performing. The difference between the two
smoothing algorithms account for the remaining dif-
ferences, with Quartz being much more permissive
in the definition of a modifiable quality value with
YALFF being more conservative.
3.2 Effects on Compression
Another criteria of evaluation is the ability of the dif-
ferent tools to improve the compressibility of quality
values. The effect on compression can be seen in last
column of Table 3.
Table 3: The compression ratios are defined as
uncompressed size
compressed size
obtained by compressing the resulting
smoothed FASTQ using
gzip
, where the uncompressed
size is 42GB. The size of the k-mer dictionary is also re-
ported.
Method Index [GB] Compression ratio
None 0 4.617
Quartz 19 6.925
YALFF hg38 5.26 7.147
YALFF Affy 5.11 7.133
YALFF dbSNP 6.27 7.241
YALFF All 6.7 7.402
First, we can observe that the processed FASTQ
file is more compressible than the original file. This
is expected as all methods tend to reduce the entropy
of quality values. The compression ratio of YALFF
is generally higher that Quartz, even if using just the
hg38 genome as reference. Moreover, if more ad-
vanced k-mer dictionaries are used, then the compres-
sion ratio increases even further.
The most evident advantage of using a com-
pressed index is the huge reduction of the dictio-
nary size meaning less RAM used during execution.
Quartz needs a powerful computer to run with large
amount of memory, at least 32GB of RAM, whereas
YALFF requires about 6-7GB of RAM, depending on
the k-mers dictionary. It must be noted that it is pos-
sible to reassemble all the unique k-mers in the hg38
reference into a more compact form. This solution
is not shown here because it is exactly equivalent in
terms of performances to the standard hg38 dictio-
nary which is more straightforward to compute, not
involving the reassembly step. The only advantage
is a reduced dimension of the k-mer dictionary from
5.26 GB to 5.09 GB.
The second advantage is the ability to store a large
number of additional variations in linear space. For
example, the total number of SNPs reported in the
popular dbSNPs and AffymetrixSNPs are about 13M.
We have shown that the hg38 and all these 13M mu-
tations can be indexed with as little as 6.7GB.
3.3 Running Time
While the reassembly of multiple k-mers into a sin-
gle linear reference allows for reduced memory con-
Indexing k-mers in Linear-space for Quality Value Compression
27

sumption and the possibility to use widespread index-
ing algorithms such as the FM-Index it also comes
with reduced query speeds. The poor locality of suc-
cinct data structures, to which the FM-Index belongs,
introduces a less efficient use of the cache. This may
impact the execution time of YALFF making it about
1.3x times slower than Quartz. This problem can
be mitigated using some optimization tricks such as
using multiple cores or extending a match on a k-
mer to search the neighbors on the linear sequence
effectively reducing the number of accesses to the
database.
4 CONCLUSIONS AND FUTURE
WORK
This work has demonstrated the feasibility of com-
bining a reassembly procedure with a string indexing
algorithm to produce a very compact dictionary of k-
mers which works as drop-in replacements whenever
static k-mer hash tables are needed. Given a SNPs
database, like dbSNPs or Affymetrix SNPs, it is pos-
sible to find a set of k-mers that are uniquely associ-
ated to a SNP. This set of k-mers can be efficiently
compressed into a string dictionary and used for qual-
ity value compression. These k-mers dictionaries are
more informative than a single reference genome and
they show better performance in terms of compres-
sion ratio and accuracy of genotyping, while keeping
low memory requirements.
Future directions of research are the construction
of a dynamic FM-Index with the ability to add and
remove k-mers without recomputing the whole struc-
ture, another interesting problem is how to speed-up
the search queries reducing cache misses. In the field
of metagenomic read classification most methods are
based on k-mers indexes (Girotto et al., 2017; Mar-
chiori and Comin, 2017; Qian et al., 2018), and only
recently the FM-index has been applied (Bˇrinda et al.,
2017), similarly the discovery of SNPs without map-
ping based on FM-index has been proposed only re-
cently (Prezza et al., 2018). We believe that the use of
FM-index will be beneficial in other alignment-free
applications like pan-genomics.
REFERENCES
Benoit, G., Lemaitre, C., Lavenier, D., Drezen, E., Dayris,
T., Uricaru, R., and Rizk, G. (2015). Reference-free
compression of high throughput sequencing data with
a probabilistic de Bruijn graph. BMC Bioinformatics,
16:288.
Bˇrinda, K. (2016). Novel computational techniques for
mapping and classifying Next-Generation Sequencing
data. PhD thesis, Universit´e Paris-Est.
Bˇrinda, K., Salikhov, K., Pignotti, S., and Kucherov, G.
(2017). Prophyle: a phylogeny-based metagenomic
classifier using the burrows-wheeler transform. Poster
at HiTSeq 2017.
Bonfield, J. K. and Mahoney, M. V. (2013). Compression
of fastq and sam format sequencing data. Plos one.
Burrows, M. and Wheeler, D. J. (1994). A block-sorting
lossless data compression algorithm. Technical report,
Digital Equipment Corporation.
Chikhi, R., Limasset, A., and Medvedev, P. (2016). Com-
pacting de bruijn graphs from sequencing data quickly
and in low memory. Bioinformatics, 32(12):i201–
i208.
C´anovas, R., Moffat, A., and Turpin, A. (2014). Lossy com-
pression of quality scores in genomic data. Bioinfor-
matics, 30(15):2130–2136.
Comin, M., Leoni, A., and Schimd, M. (2014). Qcluster:
Extending alignment-free measures with quality val-
ues for reads clustering. In Brown, D. and Morgen-
stern, B., editors, Algorithms in Bioinformatics, pages
1–13, Berlin, Heidelberg. Springer Berlin Heidelberg.
Comin, M., Leoni, A., and Schimd, M. (2015). Clustering
of reads with alignment-free measures and quality val-
ues. Algorithms for Molecular Biology, 10(1):1–10.
Consortium, T. . G. P. (2012). An integrated map of ge-
netic variation from 1,092 human genomes. Nature,
491(7422):56–65.
Ewing, B., Hillier, L., Wendl, M. C., and Green, P. (1998).
Base-Calling of Automated Sequencer Traces Using-
Phred. I. Accuracy Assessment. Genome Research,
8(3):175–185.
Ferragina, P. and Manzini, G. (2000). Opportunistic Data
Structures with Applications. In Proceedings of the
41st Annual Symposium on Foundations of Computer
Science, FOCS ’00, pages 390–, Washington, DC,
USA. IEEE Computer Society.
Ferragina, P. and Manzini, G. (2005). Indexing Compressed
Text. J. ACM, 52(4):552–581.
Girotto, S., Comin, M., and Pizzi, C. (2017). Higher re-
call in metagenomic sequence classification exploiting
overlapping reads. BMC Genomics, 18(10):917.
Girotto, S., Comin, M., and Pizzi, C. (2018a). Efficient
computation of spaced seed hashing with block index-
ing. BMC Bioinformatics, 19(15):441.
Girotto, S., Comin, M., and Pizzi, C. (2018b). Fsh: fast
spaced seed hashing exploiting adjacent hashes. Al-
gorithms for Molecular Biology, 13(1):8.
Greenfield, D. L., Stegle, O., and Rrustemi, A. (2016).
GeneCodeq: quality score compression and improved
genotyping using a Bayesian framework. Bioinfor-
matics (Oxford, England), 32(20):3124–3132.
HG38 (2018). Human reference genome (hg38). http://
hgdownload.cse.ucsc.edu/goldenpath/hg38/bigZips/.
Illumina8bin (2011). Quality scores for next-generation se-
quencing, illumina inc. Technical report, Illumina Inc.
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
28

Li, H. and Durbin, R. (2009). Fast and accurate short read
alignment with Burrows-Wheeler transform. Bioin-
formatics (Oxford, England), 25(14):1754–1760.
Li, H. and Durbin, R. (2010). Fast and accurate long-read
alignment with Burrows-Wheeler transform. Bioin-
formatics (Oxford, England), 26(5):589–595.
Malysa, G., Hernaez, M., Ochoa, I., Rao, M., Ganesan, K.,
and Weissman, T. (2015). QVZ: lossy compression
of quality values. Bioinformatics (Oxford, England),
31(19):3122–3129.
Marc¸ais, G. and Kingsford, C. (2011). A fast, lock-free
approach for efficient parallel counting of occurrences
of k-mers. Bioinformatics, 27(6):764–770.
Marchiori, D. and Comin, M. (2017). Skraken: Fast and
sensitive classification of short metagenomic reads
based on filtering uninformative k-mers. In Pro-
ceedings of the 10th International Joint Conference
on Biomedical Engineering Systems and Technologies
- Volume 3: BIOINFORMATICS, (BIOSTEC 2017),
pages 59–67. INSTICC, SciTePress.
Mohamadi, H., Chu, J., Vandervalk, B. P., and Birol, I.
(2016). nthash: recursive nucleotide hashing. Bioin-
formatics, 32(22):3492–3494.
Ochoa, I., Asnani, H., Bharadia, D., Chowdhury, M., Weiss-
man, T., and Yona, G. (2013). QualComp: a new lossy
compressor for quality scores based on rate distortion
theory. BMC bioinformatics, 14:187.
Ochoa, I., Hernaez, M., Goldfeder, R., Weissman, T., and
Ashley, E. (2017). Effect of lossy compression of
quality scores on variant calling. Briefings in Bioin-
formatics, 18(2):183–194.
Prezza, N., Pisanti, N., Sciortino, M., and Rosone, G.
(2018). Detecting Mutations by eBWT. In Parida,
L. and Ukkonen, E., editors, 18th International Work-
shop on Algorithms in Bioinformatics (WABI 2018),
volume 113 of Leibniz International Proceedings in
Informatics (LIPIcs), pages 3:1–3:15, Dagstuhl, Ger-
many. Schloss Dagstuhl–Leibniz-Zentrum fuer Infor-
matik.
ProphAsm. Compressing k-mer sets via assembling con-
tigs. https://github.com/prophyle/prophasm.
Qian, J., Marchiori, D., and Comin, M. (2018). Fast and
sensitive classification of short metagenomic reads
with skraken. Communications in Computer and In-
formation Science, 881:212–226.
Rizk, G., Lavenier, D., and Chikhi, R. (2013). Dsk: k-mer
counting with very low memory usage. Bioinformat-
ics, 29(5):652–653.
Schimd, M. and Comin, M. (2016). Fast comparison of
genomic and meta-genomic reads with alignment-free
measures based on quality values. BMC Medical Ge-
nomics, 9(1):41–50.
Shajii, A., Yorukoglu, D., William Yu, Y., and Berger,
B. (2016). Fast genotyping of known snps
through approximate k-mer matching. Bioinformat-
ics, 32(17):i538–i544.
Sherry, S. T., Ward, M.-H., Kholodov, M., Baker, J., Phan,
L., Smigielski, E. M., and Sirotkin, K. (2001). dbsnp:
the ncbi database of genetic variation. Nucleic Acids
Research, 29(1):308–311.
Shibuya, Y. and Comin, M. (2018). Better quality score
compression through sequence-based quality smooth-
ing. 15th Annual Meeting of the Bioinformatics Italian
Society. Submitted to BMC Bioinformatics.
SNPs (2018). List of known variants of the sample na12878.
ftp://ussd-ftp.illumina.com/2017-1.0/hg38.
Yu, Y. W., Yorukoglu, D., Peng, J., and Berger, B. (2015).
Quality score compression improves genotyping ac-
curacy. Nature Biotechnology, 33(3):240–243.
Zook, J. M., Chapman, B., Wang, J., Mittelman, D., Hof-
mann, O., Hide, W. A., and Salit, M. L. (2014). Inte-
grating human sequence data sets provides a resource
of benchmark snp and indel genotype calls. Nature
Biotechnology, 32:246–251.
Indexing k-mers in Linear-space for Quality Value Compression
29
