
Method Choice in Gene Set Analysis Has Important Consequences for
Analysis Outcome
Farhad Maleki
1
, Katie L. Ovens
1
, Elham Rezaei
2
, Alan M. Rosenberg
2
and Anthony J. Kusalik
1
1
Department of Computer Science, University of Saskatchewan, Saskatoon, SK, Canada
2
Department of Pediatrics, Royal University Hospital, Saskatoon, SK, Canada
Keywords:
Gene Set Analysis, Enrichment Analysis, Pathway Analysis, Gene Expression.
Abstract:
Gene set enrichment analysis is a well-established approach for gaining biological insight from expression
data. With many gene set analysis methods available, a question is raised about the consistency of the results
of these methods. In this paper, we answer this question with a systematic analysis of ten commonly used
gene set analysis methods when applied to microarray data. The statistical analysis suggests that there is a
significant difference between the results of these methods. Comparison of the 20 most statistically signifi-
cant gene sets reported by these methods showed little to no agreement regardless of the dataset being used.
This observation suggests that the outcome of a study can be highly dependent on the choice of the gene set
analysis method. Comparing the 100 most statistically significant gene sets also led to the same conclusion.
Furthermore, biological evaluation using a juvenile idiopathic arthritis dataset agreed with the results of the
statistical analysis. The 20 most statistically significant gene sets for some methods showed relevance to the
biology of juvenile arthritis, supporting their utility, while most methods led to results that were irrelevant or
marginally relevant to the known biology of the disease.
1 INTRODUCTION
High-throughput technologies have made it possible
to study the expression activity of a large number of
genes in a single experiment. These technologies are
commonly used to investigate the effect of different
stimuli on the expression activity of genes and de-
tect differential expression. A typical gene expression
study may lead to reporting several hundred genes as
being differentially expressed. Biological interpre-
tation of such an extensive list of genes is difficult.
Gene set analysis, also referred to as gene set enrich-
ment analysis, has been widely used to alleviate this
problem by detecting a concordant change in the ex-
pression pattern of groups of genes that are known to
be related to particular functions, processes, or cellu-
lar components. Such groups of genes are known as
gene sets.
Due to the lack of gold standard datasets where
the enrichment status of gene sets are a priori known,
evaluation of gene set analysis methods is challeng-
ing. In the absence of such gold standard datasets, re-
searchers have used artificial datasets to evaluate the
sensitivity and specificity of gene set analysis meth-
ods. These datasets often rely on simplifying assump-
tions about the distribution of gene expression mea-
sures. Also, they either ignore the complex gene-gene
correlation pattern among genes within gene sets or
model it using a constant value (Efron and Tibshirani,
2007; Nam and Kim, 2008; Ackermann and Strim-
mer, 2009), even though gene-gene correlation has
been reported to have a profound impact on the re-
sults of enrichment analysis methods (Tamayo et al.,
2012). Real expression datasets have also been used
to evaluate the sensitivity and specificity of gene set
analysis methods (Tarca et al., 2013). Since the true
enrichment status of gene sets are not a priori known
in real datasets, relying on unverified assumptions
about the differential enrichment of gene sets in these
datasets does not provide an authentic framework for
the evaluation of gene set analysis methods (Mathur
et al., 2018). Consequently, there is no consensus
among researchers about the method to use for a given
experimental design.
Many gene set enrichment analysis methods are
available. These methods vary in their underlying sta-
tistical model and the way they quantify a change in
the expression pattern of genes within a gene set. A
natural question that arises is whether the results of
gene set analysis are comparable across methods. In
Maleki, F., Ovens, K., Rezaei, E., Rosenberg, A. and Kusalik, A.
Method Choice in Gene Set Analysis Has Important Consequences for Analysis Outcome.
DOI: 10.5220/0007375000430054
In Proceedings of the 12th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2019), pages 43-54
ISBN: 978-989-758-353-7
Copyright
c
2019 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
43

this research, we compare the results of 10 widely
used gene set analysis methods to test if the choice
of gene set analysis method significantly affects the
result of a gene expression study. In addition, since
the most statistically significant gene sets are of more
value to researchers, we statistically and biologically
assess the agreement of the most significant gene sets
for all methods under study.
In the rest of the paper, Section 2 describes the
data and methodology used. Section 3 presents the
experimental results. The biological evaluation of the
results of gene set analysis methods are presented in
Section 4. Section 5 offers insight gained from the
experiments and provides suggestions for further re-
search. Finally, Section 6 ends the paper with a short
summary and conclusions.
2 DATA AND METHODOLOGY
2.1 Data
In this study, four large case-control experiments
in humans from the Affymetrix GeneChip Human
Genome U133 Plus 2.0 microarray platform were se-
lected for evaluation of gene set analysis methods.
These datasets originated from 1) renal cell carcinoma
tissue and healthy controls (77 controls and 77 cases,
GSE53757) (Von Roemeling et al., 2014), 2) skin
from patients with psoriasis and healthy control tis-
sue (64 controls and 58 cases, GSE13355) (Swindell
et al., 2011), 3) gingival tissues from healthy and
diseased individuals (64 controls and 183 cases,
GSE10334) (Demmer et al., 2008), and 4) blood sam-
ples from individuals with rheumatoid factor (RF)-
negative polyarthritis and healthy individuals (23 con-
trols and 35 cases, GSE26554) (Thompson et al.,
2012).
The raw data were preprocessed by first read-
ing the CEL files into R using the GEOquery ver-
sion 2.46.15 R package, and generating the normal-
ized expression table using the affy version 1.56.0
package and justRMA normalization (Irizarry et al.,
2003), which have been widely used for normalizing
Affymetrix data (Neely and Anderson, 2017; West
and Ali, 2017; Zyla et al., 2016). Probe IDs were con-
verted to their corresponding Entrez gene identifiers
using the hgu133plus2.db version 3.2.3 R package.
To avoid over-emphasizing genes with a large num-
ber of probes on the arrays, it is a common practice in
gene set analysis to collapse duplicate IDs. This was
accomplished by using collapseRows from WGCNA
version 1.61 with the MaxMean method. MaxMean
selects the probe that has the maximum average value
across samples when multiple probes map to the same
gene. Collapsing the probes resulted in 20,514 genes
in each experiment from an initial 54,675 probes.
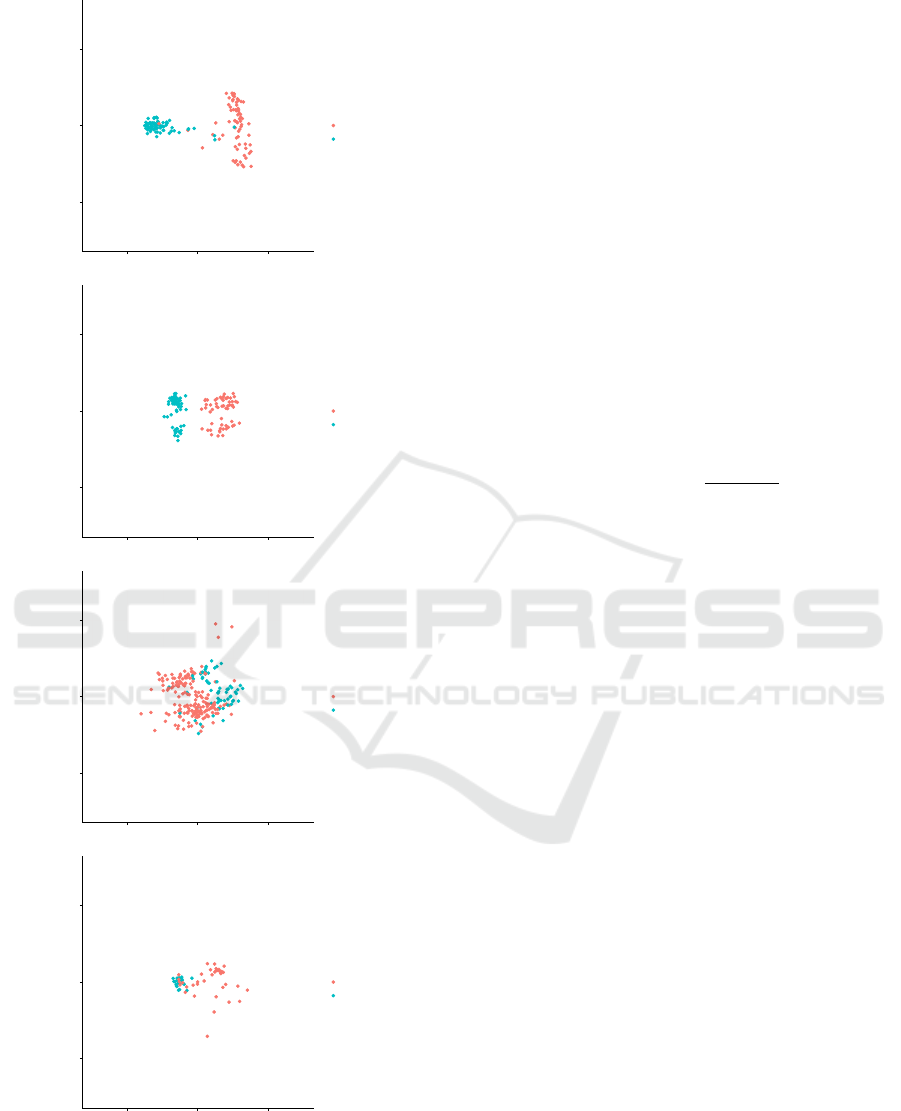
The multidimentional scaling (MDS) plots visual-
izing the case and control samples from each dataset
are shown in Figure 1. These plots were produced us-
ing cmdscale from the stats R package version 3.4.4
with default parameters.
2.2 Methodology
In this research, we compare 10 gene set anal-
ysis methods: PAGE (Kim and Volsky, 2005),
GAGE (Luo et al., 2009), Camera (Wu and Smyth,
2012), ROAST (Wu et al., 2010), FRY (from the
limma package) (Ritchie et al., 2015), GSEA (Subra-
manian et al., 2005), ssGSEA (Barbie et al., 2009),
GSVA (H
¨
anzelmann et al., 2013), PLAGE (Tom-
fohr et al., 2005), and over-representation analysis
(ORA) (Dr
˘
aghici, 2016).
The following R packages are utilized in this
study: GSVA package version 1.18.0 is used for
GSVA, PLAGE, and ssGSEA; the phyper method
from the stats package version 3.4.4 is utilized to
implement ORA; the GSEA.1.0.R script downloaded
from the Broad Institute software page for GSEA pro-
vides GSEA; the limma package version 3.34.9 is
used to run Camera, ROAST, and FRY; the gage pack-
age version 2.20.1 is used for PAGE and GAGE.
In addition to a gene expression dataset, gene set
analysis requires a database of gene sets as input. In
this research, we used the GO gene sets—hereafter
referred to as G—extracted from MSigDB version
6.1 (Subramanian et al., 2005). The GO database is
widely used for gene set analysis.
For each gene expression dataset D
i
and method
ψ
j
, gene set analysis is conducted using the default
parameters proposed by the authors of ψ
j
. To adjust
for multiple comparisons, the Benjamini-Hochberg
adjustment (Benjamini and Hochberg, 1995) with a
false discovery rate of 0.05 is applied. The resulting
adjusted p-values are denoted by a vector R
ψ
j
D
i
, where
R
ψ
j
D
i
(n)—the n
th
element of this vector—represents
the adjusted p-value resulting from gene set analysis
of the n
th
gene set in the gene set database G using
method ψ
j
.
For a significance level α = 0.05, we define a vec-
tor E
ψ
j
D
i
as follows:
E
ψ
j
D
i
(n) =
(
1, if R
ψ
j
D
i
(n) < α
0, otherwise
(1)
where E
ψ
j
D
i
represents the predicted differential en-
richment status of gene sets in G—1 for differentially
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
44
−100
0
100
−100 0 100
Case
Control
GSE53757
−100
0
100
−100 0 100
Case
Control
GSE13355
−100
0
100
−100 0 100
Case
Control
GSE10334
−100
0
100
−100 0 100
Case
Control
GSE26554
Figure 1: MDS plots for samples from the datasets un-
der study. The MDS plots from top to bottom are for
datasets with GEO IDs GSE35757, GSE13355, GSE10334,
and GSE26554, respectively.
enriched and 0 for non-differentially enriched—and
E
ψ
j
D
i
(n) is the n
th
element of E
ψ
j
D
i
. This is accom-
plished using cochran.qtest method from the RVAide-
Memoire R package version 0.9.69.3.
For a given dataset D
i
, we statistically assess
whether there is a significant difference between these
predictions across different methods or not. Since
the enrichment status is a dichotomous variable and
there are paired data for each gene set (enrichment
status for the same gene set across methods), we con-
duct Cochran’s Q test for E
ψ
j
D
i
across all values of ψ
j
,
i.e. all methods. As post hoc analysis, Wilcoxon
sign test is conducted for pairwise comparisons if
the Cochran’s Q test suggests a significant difference
across methods.
Moreover, given the result of two gene set analysis
methods ψ
j
and ψ
k
, we compare the similarity of their
results when analyzing dataset D
i
using the Jaccard
index (Bakus, 2007) as follows:
J(S
ψ
j
D
i
, S
ψ
k
D
i
) =
S
ψ
j
D
i
∩ S
ψ
k
D
i
S
ψ
j
D
i
∪ S
ψ
k
D
i
(2)
where S
ψ
j
D
i
is the set of all statistically significant gene
sets—i.e. gene sets with an adjusted p-value less than
α—when analyzing dataset D
i
using ψ
j
. A Jaccard
index of 1 corresponds to the highest similarity, i.e.
S
ψ
j
D
i
= S
ψ
k
D
i
, while a Jaccard index of 0 represents no
similarity. Also, we define the Jaccard index to be 1
if S
ψ
j
D
i
and S
ψ
k
D
i
both are empty sets. In this paper, we
interchangeably refer to the Jaccard index as overlap
score.
In addition, since the most statistically significant
results—i.e. gene sets predicted as being differen-
tially enriched with the lowest p-values—are of the
most interest to researchers, we investigate the agree-
ment among the methods regarding their most signif-
icant results. In this regard, we define S (D
i
, ψ
j
,t)
to be the set of up to t statistically most significant
gene sets predicted as being differentially enriched—
with an adjusted p-value less than α—when analyzing
dataset D
i
using ψ
j
. It should be noted that in cases
where the number of differentially enriched gene sets
is less than t, S (D
i
, ψ
j
,t) is equal to the entire set of
differentially enriched gene sets resulting from analy-
sis of D
i
using ψ
j
. After determining S (D
i
, ψ
j
,t) for
each method ψ
j
, we quantify the agreement of differ-
ent methods for their most significant results using an
overlap score of J(S (D
i
, ψ
j
,t) , S (D
i
, ψ
k
,t)). In this
research, we investigate agreement between the top
20 (and also the top 100) most significant results re-
ported by each method.
Method Choice in Gene Set Analysis Has Important Consequences for Analysis Outcome
45

3 EXPERIMENTAL RESULTS
First, each of the four datasets was analyzed using the
ten methods under study. Next, E
ψ
j
D
i
, i.e. the differen-
tial enrichment status of gene sets in G, were deter-
mined. For each dataset D
i
a Cochran’s Q test with
a significance level α = 0.05 was used to statistically
assess if there is a significant difference between the
differential enrichment status of gene sets in G across
the methods under study. The Cochran’s Q test for all
datasets showed a statistically significant difference
between the results of the methods under study (see
Tables 2 to 6 in the Appendix for test results and the
post hoc analysis).
Figure 2, using a series of triangular heat maps,
illustrates the extent of overlap between the results of
the 10 gene set analysis methods for the four datasets
and three different scenarios: 1) when overlap is mea-
sured from the top 20 most significant gene sets pre-
dicted by each method, 2) when overlap is measured
from the top 100 most significant gene sets predicted
by each method, and 3) when overlap is measured
from all the significant gene sets predicted by each
method. Each cell in these heat maps represents the
overlap score between the results of two methods. A
blue hue of a cell indicates low overlap and a red hue
indicates high overlap in enriched gene sets between
two methods. The heat maps in Figure 2 show that, re-
gardless of the datasets being used, the consistency—
as measured by overlap score—between the results
of different gene set analysis methods is generally
low. However, as we move from scenario 1 to 3, the
overlap between the results of some of the methods
increases. In some instances, such as ROAST and
FRY, the amount of overlap remains consistently high
across scenarios. The consistency among methods
when considering the top 20 most statistically signifi-
cant results is much lower than the consistency when
considering all significant results. This pattern is also
observed when comparing the top 100 most statisti-
cally significant gene sets to all gene sets predicted
as being differentially enriched. Also, Camera and
GSEA have little consistency with all other methods
under study.
Table 1 shows the total number of differentially
enriched gene sets reported for all the datasets and
all ten methods. GSEA, Camera, and ORA predict a
smaller number of gene sets as differentially enriched
compared to the other methods.
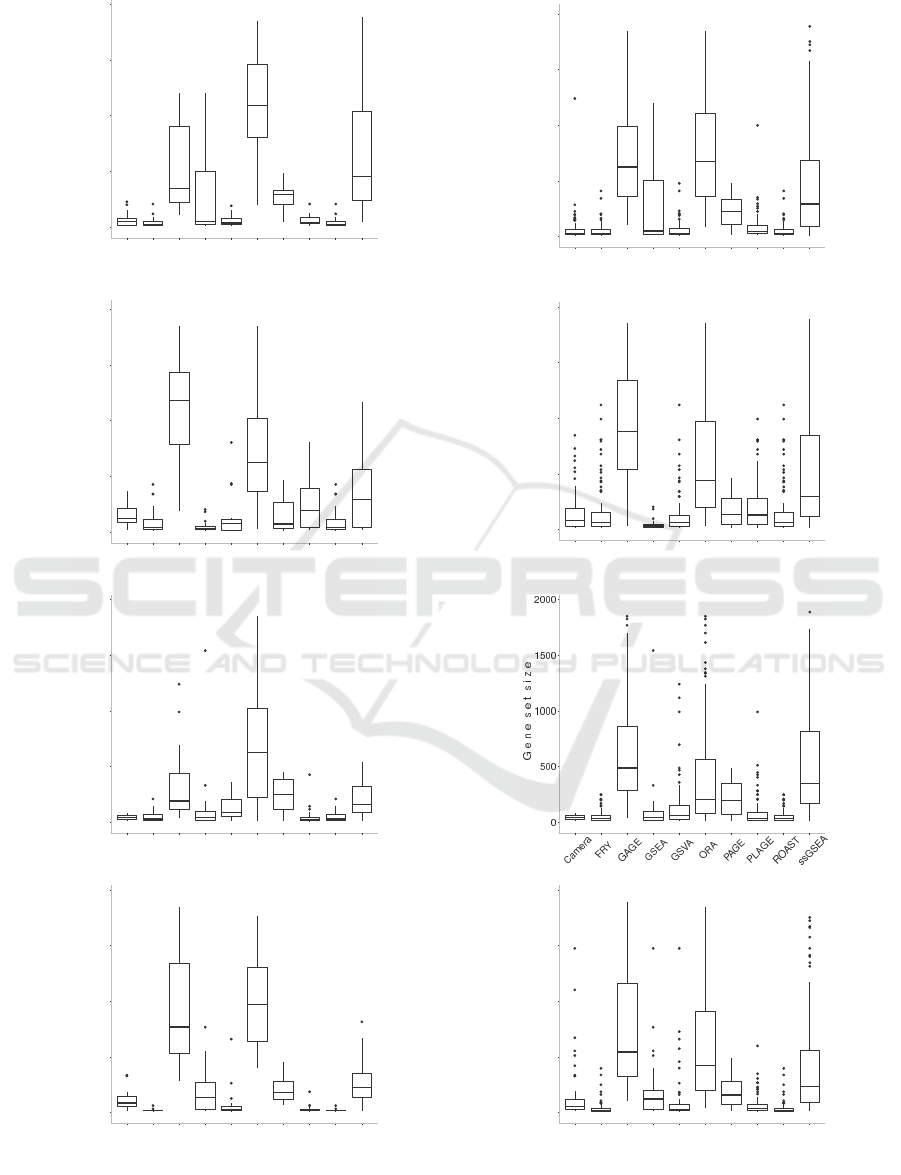
Figure 3 visualizes the distribution of the size of
the top 20 and the top 100 most significant gene
sets predicted as being differentially enriched for each
method. These box plots further highlight the differ-
ence between the results of the gene set analysis meth-
ods. GAGE, ORA, and ssGSEA tend to report larger
gene sets, i.e. gene sets that contain higher numbers
of genes, in comparison to the other methods regard-
less of the dataset being analyzed.
Table 1: Number of gene sets predicted as being differen-
tially enriched by each method for each dataset.
GSE53757 GSE13355 GSE10334 GSE26554
FRY 4937 4876 4241 3660
GSEA 19 17 17 33
ORA 1547 573 130 222
Camera 155 73 3 313
ssGSEA 5844 5869 5862 5846
PAGE 1967 1400 1375 1054
GSVA 4730 3847 3819 2988
PLAGE 5900 5830 5242 5698
ROAST 4949 4737 4256 3380
GAGE 3951 3899 3887 2441
4 BIOLOGICAL EVALUATION
Juvenile idiopathic arthritis (JIA) is a class of child-
hood arthritis with unknown cause developing be-
fore the age of 16 years and persisting for at least
6 weeks. JIA comprises seven categories including:
1) systemic arthritis, 2) oligoarthritis, 3) polyarthri-
tis rheumatoid factor (RF)-negative, 4) polyarthri-
tis RF-positive, 5) psoriatic arthritis, 6) enthesitis-
related arthritis (ERA), and 7) undifferentiated (Petty
et al., 2004). For biological validation of methods
under study, a JIA dataset containing RF-negative
polyarthritis samples and healthy controls was ob-
tained from the same Affymetrix GeneChip Human
Genome U133 Plus 2.0 microarray platform as the
other datasets (23 controls and 35 cases, GSE26554).
Expression profiles tend to be distinguishable
among JIA categories. Gene expression and
genome-wide genotyping have identified genes as-
sociated with different JIA subtypes, particularly
HLA gene complex, PTPN22, PTPN2, STAT4,
ANKRD55, Interleukin (IL)2-IL21, IL-2RA, IL-6,
SH2B3-ATXN2, MIF, SLC11A1 (NRAMP1), TNFA,
TNFAIP3, TRAF1/C5, VTCN1, CCL5, CD14, and
WISP3 (Prahalad, 2004; Phelan et al., 2006; Praha-
lad and Glass, 2008; Martinez et al., 2008; Yao et al.,
2009; Fung et al., 2009). The functions of these
genes are chiefly regulating production and function
of inflammatory biomarkers and their receptors. For
instance, PTPN2 modulates the expression of IL-2,
IL-4, IL-6, and IFN. Variants of this gene can cause
impairment in the regulation of inflammatory path-
ways, including joint inflammation (Jorde, 2000; Pra-
halad, 2006; Prahalad et al., 2000). The inflammatory
process is mediated by an array of innate regulators
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
46

Top 20 results Top 100 results All results
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
FRY
GSEA
ORA
Camera
ssGSEA
PAGE
GSVA
PLAGE
ROAST
GAGE
GSE53757
GSE13355
GSE10334
GSE26554
0.00 0.25 0.50 0.75 1.00
Overlap Score
Figure 2: A set of triangular heat maps depicting the consistency of the results of gene set analysis methods—as measured
by overlap score—across databases. Each triangular heat map illustrates the overlap score of the results of gene set analysis
methods when analyzing a gene expression dataset. The layers in the plot, from top to bottom, correspond to datasets with
GEO id of GSE53757, GSE13355, GSE10334, and GSE26554, respectively. Ranging from 0 to 1, the overlap score is
represented by color hues from blue to red, separated by yellow in the middle (overlap of 0.5). The plot suggests that there
is little consistency between the results of the gene set analysis methods under study. This lack of consistency is more
pronounced among the top 20 (left column) and top 100 (middle column) most statistically significant results compared to all
differentially enriched gene sets (right column).
including interleukins, chemokines, growth factors,
and matrix metalloproteinases (MMPs)(Petty et al.,
2015). There has been increasing interest in iden-
tifying molecules involved in regulating immune re-
sponses related to susceptibility to, and outcome of,
JIA.
Biological evaluation of the 10 gene set enrich-
ment analysis methods under study was performed
based on the gene sets/pathways that are known to
play a role in JIA using the dataset GSE26554 to de-
termine the biological relevance of the gene sets pre-
dicted as being differentially enriched.
All of the top 20 gene sets predicted as being dif-
ferentially enriched by GAGE showed general rel-
evance to JIA. For example, the top 3 gene sets
were “immune response” (GO:0006955), “regulation
Method Choice in Gene Set Analysis Has Important Consequences for Analysis Outcome
47
Top 20 most significant results Top 100 most significant results
0
500
1000
1500
2000
Camera
FRY
GAGE
GSEA
GSVA
ORA
PAGE
PLAGE
ROAST
ssGSEA
G e n e s e t s i z e
GSE53757
0
500
1000
1500
2000
Camera
FRY
GAGE
GSEA
GSVA
ORA
PAGE
PLAGE
ROAST
ssGSEA
G e n e s e t s i z e
GSE53757
0
500
1000
1500
2000
Camera
FRY
GAGE
GSEA
GSVA
ORA
PAGE
PLAGE
ROAST
ssGSEA
G e n e s e t s i z e
GSE13355
0
500
1000
1500
2000
Camera
FRY
GAGE
GSEA
GSVA
ORA
PAGE
PLAGE
ROAST
ssGSEA
G e n e s e t s i z e
GSE13355
0
500
1000
1500
2000
Camera
FRY
GAGE
GSEA
GSVA
ORA
PAGE
PLAGE
ROAST
ssGSEA
G e n e s e t s i z e
GSE10334
GSE10334
0
500
1000
1500
2000
Camera
FRY
GAGE
GSEA
GSVA
ORA
PAGE
PLAGE
ROAST
ssGSEA
G e n e s e t s i z e
GSE26554
0
500
1000
1500
2000
Camera
FRY
GAGE
GSEA
GSVA
ORA
PAGE
PLAGE
ROAST
ssGSEA
G e n e s e t s i z e
GSE26554
Figure 3: Box plots visualizing the distribution of gene set size among the top 20 (left) and top 100 (right) most statisti-
cally significant gene sets reported by each method. The plots, from top to bottom, correspond to datasets with GEO id of
GSE53757, GSE13355, GSE10334, and GSE26554 respectively.
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
48

of immune system process” (GO:0002682), and “im-
mune system process” (GO:0002376). All these gene
sets are relatively large and nonspecific to the ac-
tual disease or related pathways, with sizes approach-
ing or exceeding 1000 genes. Several gene sets con-
tained fewer genes, while still potentially relating to
JIA, including “response to cytokine” (GO:0034097)
and “inflammatory response” (GO:0006954). Fur-
thermore, many of the gene sets in the top 20 pre-
dicted using GAGE had many terms relating to a gen-
eral immune process or response (8 of the top 20 gene
sets predicted as being differentially enriched).
GSEA also predicted a moderate number of gene
sets that are thought to play a role in JIA, but unlike
GAGE, these gene sets were much smaller and re-
lated to more specific processes. The small predicted
gene sets related to JIA included the HLA complex
in the gene set “trans-Golgi network” (GO:0005802).
Other gene sets predicted as differentially enriched
included “positive regulation of antigen receptor-
mediated signaling pathway” (GO:0050857), which
involves the cross-linking of antigen receptors of im-
mune cells, and “cellular response to interferon-beta”
(GO:0035458), which involves responses to a partic-
ular cytokine.
ORA also predicted a moderate number of gene
sets related to cytokines, while PAGE predicted gene
sets related to immune response. The few gene sets
relevant to JIA reported by ORA and PAGE were also
reported by GAGE. This agrees with our observations
in Section 3, as the results of these three methods
moderately overlap. The other six methods produced
few gene sets—among their top results—associated
with immune response or inflammation, as shown by
the overlap scores (in the triangular heat maps for
dataset GSE26554) for the top 20 and top 100 most
significant results reported by these methods.
5 DISCUSSION
In this research, we showed that there is a significant
difference between the results of ten commonly used
gene set analysis methods. We quantified the similar-
ity between the results of the methods using Jaccard
index. Since researchers value the most statistically
significant results, we studied the distribution of gene
set size for the top 20 and top 100 most significant
results of each method.
The results showed that ROAST and FRY share
the same top 20 and top 100 significantly enriched
gene sets. This is expected as FRY was designed to be
a computationally efficient approximation of ROAST.
Also, there are moderate overlaps between the top
20 (and top 100) most significant results reported by
ORA, GAGE, and PAGE. This similarity can be ex-
plained as all three of these methods are paramet-
ric gene set analysis methods; ORA and GAGE are
based on two-sample t-tests, and PAGE is based on
a z-score. These observations support the validity of
the experimental design in this research.
When considering all gene sets predicted as be-
ing differentially enriched, there are moderate to high
overlaps between the results of all methods, with the
exception of ORA, Camera, and GSEA. These high
overlaps appear to be a consequence of the high num-
ber of gene sets reported as being differentially en-
riched. As seen by combining the results in Figure 2
and Table 1, two methods with high overlap between
their results also report a high number of gene sets
as being differentially enriched. This happens when
some methods report a large proportion of gene sets
as being differentially enriched. At the extreme, if
two methods report all gene sets, the overlap will be
its maximum value, i.e. 1. However, as also depicted
in Figure 2, there is no or very small overlap between
the top 20 (and top 100) differentially enriched gene
sets reported by methods that achieve high overlap
scores when considering all of their significant re-
sults. These observations suggest that the methods
under study generally do not agree in the gene sets
they reported as most statistically significant.
The high numbers of reported differentially en-
riched gene sets (for some methods) are not an ar-
tifact of the choice of the expression datasets or the
preprocessing steps. Single gene expression analy-
sis reported that GSE53757 had 571 differentially ex-
pressed genes, GSE13355 had 121, GSE10334 had
12, and GSE26554 had 5 differentially expressed
genes. These results were produced using the limma
package with a log fold change cutoff of ±2, a
Benjamini-Hochberg correction for multiple compar-
isons, and a significant level α = 0.05.
The number of differentially enriched gene sets re-
ported by ORA and Camera, compared to all other
methods under study, seem to be more sensitive to the
variation between the case and control groups of each
dataset (see Figure 1). For the datasets that have more
distinct groups, more gene sets are reported as differ-
entially enriched. When the variation is low, ORA—
for example—predicts fewer differentially enriched
gene sets. One explanation is that the list of genes
predicted as being differentially expressed, an input to
ORA, is based on a t-test. The t-test statistic denom-
inator represents the variance of expression measures
for a gene, and when the sample variation is high, as
with GSE10334, the statistic value decreases and the
derived p-value increases; this results in a small num-
Method Choice in Gene Set Analysis Has Important Consequences for Analysis Outcome
49

ber of differentially expressed genes. This, in turn,
decreases the number of differentially enriched gene
sets reported by ORA.
GSEA and Camera typically report a small num-
ber of differentially enriched gene sets for each data
set. Since the number of reported differentially en-
riched gene sets is small, these methods have a very
small overlap with the results of other methods and
also to each other.
Gene sets extracted from GO are associated with
GO terms, where the more general terms usually cor-
respond to larger gene sets, and specific terms corre-
spond to smaller gene sets. As depicted in Figure 3,
for the 20—and also top 100—most statistically sig-
nificant gene sets reported, ORA, GAGE, PAGE, and
ssGSEA tend to report gene sets with larger sizes. Al-
though there could be cases where a gene set with a
large size is associated with a phenotype of interest,
these three methods consistently report larger gene
sets across the datasets compared to the other meth-
ods. This may be a sign of systematic bias in favour
of large gene set sizes, which are usually less informa-
tive. With ORA in particular, the amount of variation
between gene set sizes tends to be high as well; also,
the median gene set size is typically higher than all
other methods. On the other hand, reported gene sets
by GSEA have a low median size, although variation
between sizes is larger than some of the other meth-
ods such as Camera, FRY, ROAST, and GSVA. This
is because GSEA reports a mixture of small gene sets
followed by large gene sets in the top 20 and 100 re-
sults. Camera also reports a small number of gene
sets, usually with small sizes. PLAGE, FRY, ROAST,
and GSVA—on the other hand—report a large num-
ber of gene sets as differentially enriched but, like
Camera, their most significant gene sets have small
sizes. This could suggest that the reported gene sets
are very specific to a particular biological process,
molecular function, or cellular component. However,
this does not necessarily mean these gene sets are bi-
ologically informative to the phenotype under study.
To assist with interpreting these results, the relevancy
of the most significant genes sets for the JIA dataset
was explored by biological interpretation.
The biological evaluation in Section 4 suggests
that GAGE performed the best followed by GSEA,
ORA, and PAGE at predicting the most gene sets that
were relevant to the phenotype of interest. This is
on par with the overlap scores where GAGE achieved
moderate overlap scores with ORA and PAGE. GSEA
reports a small number of gene sets as being differ-
entially enriched and therefore achieves low overlap
scores with other methods. However, some of its re-
ported gene sets showed relevance to specific immune
system processes, which could be more informative
compared to some of the more general gene sets re-
ported by GAGE, ORA, and PAGE. We suggest that
these results be confirmed further with validation per-
formed on a wide variety of datasets to ensure the re-
sults are not dataset or phenotype dependent.
These observations further highlight the lack of
agreement between the results of gene set analysis
methods. Our results support the utility of methods
such as GAGE, GSEA, ORA, and PAGE in gaining
biological insight. Drawing a conclusion based on the
results of the other methods, even their most signifi-
cant results, is more challenging and prone to inves-
tigator bias toward a hypothesis of interest. This is
even a more serious problem for methods that report
a large number of gene sets as being differentially en-
riched. Since it is unlikely for a living organism to un-
dergo such a dramatic change involving several thou-
sand gene sets, this can be interpreted as the lack of
specificity, i.e. incorrectly reporting a large number
of gene sets as being differentially enriched. We sug-
gest developing methods with higher specificity with-
out sacrificing sensitivity as future research.
Often, it is the case that researchers studying the
same phenomenon come up with different results (e.g.
different implicated gene sets) even though they ap-
pear to have each followed a valid methodology. We
are left searching for an explanation for the difference
in results. Since our study shows that there is a lack
of consistency between the results of gene set anal-
ysis methods, part of the explanation could be using
different gene set analysis methods, if different gene
set analysis methods were used.
6 CONCLUSION
In this paper, we studied the consistency of the re-
sults of ten commonly used gene set analysis methods
when applied to real expression datasets. The data
analysis showed that there is a significant difference
between the results of these methods. Our study sug-
gests that not only do these methods differ in the gene
sets reported as being differentially enriched, but they
also differ in the distribution of the size of the reported
gene sets. Further, there is little to no overlap between
the results of top 20 (and top 100) most statistically
significant gene sets reported, except between FRY
and ROAST.
The biological validation of the most significant
results using a JIA dataset revealed that GAGE per-
forms the best followed by GSEA, ORA, and PAGE
at predicting the most gene sets relevant to the phe-
notype of interest. The biological evaluation of the
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
50

most significant results reported by the gene set analy-
sis methods revealed that the majority of the methods
reported gene sets that are not related to the known
biology of JIA. GAGE was the only method with all
of its top 20 gene sets relevant to the biology of juve-
nile arthritis. In addition, GSEA, ORA, and PAGE re-
ported relevant gene sets, with GSEA reporting fewer
but more specific gene sets. This supports the util-
ity of these methods for gene set analysis. However,
any more general conclusion would require a broader
study.
REFERENCES
Ackermann, M. and Strimmer, K. (2009). A general modu-
lar framework for gene set enrichment analysis. BMC
Bioinformatics, 10(1):47.
Bakus, G. J. (2007). Quantitative analysis of marine bio-
logical communities: field biology and environment.
John Wiley & Sons.
Barbie, D. A., Tamayo, P., Boehm, J. S., Kim, S. Y., Moody,
S. E., Dunn, I. F., Schinzel, A. C., Sandy, P., Meylan,
E., Scholl, C., et al. (2009). Systematic RNA inter-
ference reveals that oncogenic kras-driven cancers re-
quire tbk1. Nature, 462(7269):108.
Benjamini, Y. and Hochberg, Y. (1995). Controlling the
false discovery rate: a practical and powerful ap-
proach to multiple testing. Journal of the Royal Sta-
tistical Society: Series B (Statistical Methodology),
57(1):289–300.
Demmer, R. T., Behle, J. H., Wolf, D. L., Handfield, M.,
Kebschull, M., Celenti, R., Pavlidis, P., and Papa-
panou, P. N. (2008). Transcriptomes in healthy and
diseased gingival tissues. Journal of Periodontology,
79(11):2112–2124.
Dr
˘
aghici, S. (2016). Statistics and data analysis for mi-
croarrays using R and bioconductor. CRC Press.
Efron, B. and Tibshirani, R. (2007). On testing the signifi-
cance of sets of genes. The Annals of Applied Statis-
tics, 1(1):107–129.
Fung, E., Smyth, D., Howson, J., Cooper, J., Walker, N.,
Stevens, H., Wicker, L., and Todd, J. (2009). Analysis
of 17 autoimmune disease-associated variants in type
1 diabetes identifies 6q23/tnfaip3 as a susceptibility
locus. Genes and immunity, 10(2):188.
H
¨
anzelmann, S., Castelo, R., and Guinney, J. (2013).
GSVA: gene set variation analysis for microarray and
RNA-seq data. BMC Bioinformatics, 14(1):7.
Irizarry, R. A., Bolstad, B. M., Collin, F., Cope, L. M.,
Hobbs, B., and Speed, T. P. (2003). Summaries of
affymetrix genechip probe level data. Nucleic Acids
Research, 31(4):e15–e15.
Jorde, L. (2000). Linkage disequilibrium and the search
for complex disease genes. Genome research,
10(10):1435–1444.
Kim, S.-Y. and Volsky, D. J. (2005). PAGE: parametric
analysis of gene set enrichment. BMC Bioinformat-
ics, 6(1):144.
Luo, W., Friedman, M. S., Shedden, K., Hankenson, K. D.,
and Woolf, P. J. (2009). GAGE: generally applica-
ble gene set enrichment for pathway analysis. BMC
Bioinformatics, 10(1):161.
Martinez, A., Varade, J., Marquez, A., Cenit, M., Espino,
L., Perdigones, N., Santiago, J., Fern
´
andez-Arquero,
M., De La Calle, H., Arroyo, R., et al. (2008). As-
sociation of the stat4 gene with increased susceptibil-
ity for some immune-mediated diseases. Arthritis &
Rheumatism, 58(9):2598–2602.
Mathur, R., Rotroff, D., Ma, J., Shojaie, A., and Motsinger-
Reif, A. (2018). Gene set analysis methods: a system-
atic comparison. BioData Mining, 11(1):8.
Nam, D. and Kim, S.-Y. (2008). Gene-set approach for ex-
pression pattern analysis. Briefings in Bioinformatics,
9(3):189–197.
Neely, B. A. and Anderson, P. E. (2017). Complementary
domain prioritization: A method to improve biolog-
ically relevant detection in multi-omic data sets. In
Proceedings of the 10th International Joint Confer-
ence on Biomedical Engineering Systems and Tech-
nologies - Volume 3: BIOINFORMATICS, (BIOSTEC
2017), pages 68–80. INSTICC, SciTePress.
Petty, R., Laxer, R., Lindsley, C., and Wedderburn, L.
(2015). Textbook of Pediatric Rheumatology. Elsevier
Health Sciences.
Petty, R. E., Southwood, T. R., Manners, P., Baum, J., Glass,
D. N., Goldenberg, J., He, X., Maldonado-Cocco, J.,
Orozco-Alcala, J., Prieur, A.-M., et al. (2004). Inter-
national league of associations for rheumatology clas-
sification of juvenile idiopathic arthritis: second revi-
sion, edmonton, 2001. The Journal of rheumatology,
31(2):390.
Phelan, J., Thompson, S., and Glass, D. (2006). Suscepti-
bility to jra/jia: complementing general autoimmune
and arthritis traits. Genes and immunity, 7(1):1.
Prahalad, S. (2004). Genetics of juvenile idiopathic arthri-
tis: an update. Current opinion in rheumatology,
16(5):588–594.
Prahalad, S. (2006). Genetic analysis of juvenile rheuma-
toid arthritis: approaches to complex traits. Cur-
rent problems in pediatric and adolescent health care,
36(3):83.
Prahalad, S. and Glass, D. N. (2008). A comprehensive
review of the genetics of juvenile idiopathic arthritis.
Pediatric Rheumatology, 6(1):11.
Prahalad, S., Ryan, M. H., Shear, E. S., Thompson, S. D.,
Giannini, E. H., and Glass, D. N. (2000). Juve-
nile rheumatoid arthritis: linkage to hla demonstrated
by allele sharing in affected sibpairs. Arthritis &
Rheumatism: Official Journal of the American Col-
lege of Rheumatology, 43(10):2335–2338.
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi,
W., and Smyth, G. K. (2015). limma powers differen-
tial expression analyses for RNA-sequencing and mi-
croarray studies. Nucleic Acids Research, 43(7):e47–
e47.
Method Choice in Gene Set Analysis Has Important Consequences for Analysis Outcome
51

Subramanian, A., Tamayo, P., Mootha, V. K., Mukher-
jee, S., Ebert, B. L., Gillette, M. A., Paulovich, A.,
Pomeroy, S. L., Golub, T. R., Lander, E. S., et al.
(2005). Gene set enrichment analysis: a knowledge-
based approach for interpreting genome-wide expres-
sion profiles. PNAS, 102(43):15545–15550.
Swindell, W. R., Johnston, A., Carbajal, S., Han, G., Wohn,
C., Lu, J., Xing, X., Nair, R. P., Voorhees, J. J., Elder,
J. T., et al. (2011). Genome-wide expression profiling
of five mouse models identifies similarities and differ-
ences with human psoriasis. PloS One, 6(4):e18266.
Tamayo, P., Steinhardt, G., Liberzon, A., and Mesirov, J. P.
(2012). The limitations of simple gene set enrich-
ment analysis assuming gene independence. Statis-
tical Methods in Medical Research, 25(1):1–16.
Tarca, A. L., Bhatti, G., and Romero, R. (2013). A
comparison of gene set analysis methods in terms of
sensitivity, prioritization and specificity. PloS One,
8(11):e79217.
Thompson, S. D., Marion, M. C., Sudman, M., Ryan,
M., Tsoras, M., Howard, T. D., Barnes, M. G.,
Ramos, P. S., Thomson, W., Hinks, A., et al. (2012).
Genome-wide association analysis of juvenile idio-
pathic arthritis identifies a new susceptibility locus at
chromosomal region 3q13. Arthritis & Rheumatism,
64(8):2781–2791.
Tomfohr, J., Lu, J., and Kepler, T. B. (2005). Pathway level
analysis of gene expression using singular value de-
composition. BMC Bioinformatics, 6(1):225.
Von Roemeling, C. A., Radisky, D. C., Marlow, L. A.,
Cooper, S. J., Grebe, S. K., Anastasiadis, P. Z., Tun,
H. W., and Copland, J. A. (2014). Neuronal pentraxin
2 supports clear cell renal cell carcinoma by activat-
ing the ampa-selective glutamate receptor-4. Cancer
Research, 74(17):4796–4810.
West, S. and Ali, H. (2017). Sensitivity analysis of granu-
larity levels in complex biological networks. In Fred,
A. and Gamboa, H., editors, Biomedical Engineering
Systems and Technologies, pages 167–188. Springer
International Publishing.
Wu, D., Lim, E., Vaillant, F., Asselin-Labat, M.-L., Vis-
vader, J. E., and Smyth, G. K. (2010). ROAST: ro-
tation gene set tests for complex microarray experi-
ments. Bioinformatics, 26(17):2176–2182.
Wu, D. and Smyth, G. K. (2012). Camera: a competitive
gene set test accounting for inter-gene correlation. Nu-
cleic Acids Research, 40(17):e133–e133.
Yao, T.-C., Tsai, Y.-C., and Huang, J.-L. (2009). Associ-
ation of rantes promoter polymorphism with juvenile
rheumatoid arthritis. Arthritis & Rheumatism: Offi-
cial Journal of the American College of Rheumatol-
ogy, 60(4):1173–1178.
Zyla, J., Marczyk, M., and Polanska, J. (2016). Sensitiv-
ity, specificity and prioritization of gene set analysis
when applying different ranking metrics. In 10th In-
ternational Conference on Practical Applications of
Computational Biology & Bioinformatics, pages 61–
69. Springer.
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
52

APPENDIX
Table 2: The results of Cochran’s Q test for all datasets.
Dataset Q Statistic
Degrees of
freedom
p-value
GSE53757 32133.2 9 <2.2e-16
?
GSE13355 33592.8 9 <2.2e-16
?
GSE10334 31661.4 9 <2.2e-16
?
GSE26554 29326.9 9 <2.2e-16
?
?
2.2e-16 is the smallest p-value reported by cochran.qtest method from RVAideMemoire
Table 3: The p-values of Wilcoxon sign tests for pairwise comparisons of the results of the methods using dataset GSE53757.
Camera FRY GAGE GSEA GSVA ORA PAGE PLAGE ROAST
FRY 4.94e-324
GAGE 0.00e+00 2.51e-101
GSEA 4.47e-28 0.00e+00 0.00e+00
GSVA 0.00e+00 2.37e-09 1.46e-57 0.00e+00
ORA 0.00e+00 0.00e+00 0.00e+00 0.00e+00 0.00e+00
PAGE 0.00e+00 0.00e+00 0.00e+00 0.00e+00 0.00e+00 1.62e-44
PLAGE 0.00e+00 5.21e-282 0.00e+00 0.00e+00 0.00e+00 4.94e-324 4.94e-324
ROAST 0.00e+00 4.41e-01 4.69e-103 0.00e+00 2.47e-10 0.00e+00 0.00e+00 9.51e-277
ssGSEA 4.94e-324 5.41e-207 0.00e+00 0.00e+00 1.15e-266 0.00e+00 0.00e+00 2.04e-09 1.57e-202
Note that a p-value of 0.00e+00 is smaller than the representable floating-point precision.
Table 4: The p-values of Wilcoxon sign tests for pairwise comparisons of the results of the methods using dataset GSE13355.
Camera FRY GAGE GSEA GSVA ORA PAGE PLAGE ROAST
FRY 0.00e+00
GAGE 0.00e+00 4.08e-104
GSEA 1.25e-09 9.88e-324 0.00e+00
GSVA 0.00e+00 4.66e-159 3.03e-01 0.00e+00
ORA 2.78e-101 9.88e-324 0.00e+00 1.05e-152 0.00e+00
PAGE 0.00e+00 0.00e+00 0.00e+00 0.00e+00 0.00e+00 9.55e-152
PLAGE 9.88e-324 6.49e-233 0.00e+00 9.88e-324 0.00e+00 0.00e+00 0.00e+00
ROAST 0.00e+00 5.33e-15 6.33e-74 0.00e+00 5.39e-134 0.00e+00 0.00e+00 4.37e-272
ssGSEA 0.00e+00 7.62e-253 0.00e+00 0.00e+00 0.00e+00 0.00e+00 0.00e+00 1.02e-03 1.81e-295
Note that a p-value of 0.00e+00 is smaller than the representable floating-point precision.
Table 5: The p-values of Wilcoxon sign tests for pairwise comparisons of the results of the methods using dataset GSE10334.
Camera Camera FRY GAGE GSEA GSVA ORA PAGE PLAGE ROAST
FRY 0.00e+00
GAGE 0.00e+00 1.55e-11
GSEA 2.70e-03 4.94e-324 0.00e+00
GSVA 0.00e+00 2.36e-100 2.04e-01 0.00e+00
ORA 4.41e-37 4.94e-324 0.00e+00 4.44e-23 4.94e-324
PAGE 0.00e+00 4.94e-324 0.00e+00 0.00e+00 0.00e+00 0.00e+00
PLAGE 0.00e+00 9.91e-132 6.79e-212 4.94e-324 3.28e-229 0.00e+00 0.00e+00
ROAST 0.00e+00 5.00e-02 1.93e-12 0.00e+00 1.70e-104 0.00e+00 4.94e-324 7.23e-128
ssGSEA 0.00e+00 0.00e+00 0.00e+00 4.94e-324 0.00e+00 4.94e-324 4.94e-324 6.10e-145 0.00e+00
Note that a p-value of 0.00e+00 is smaller than the representable floating-point precision.
Method Choice in Gene Set Analysis Has Important Consequences for Analysis Outcome
53

Table 6: The p-values of Wilcoxon sign tests for pairwise comparisons of the results of the methods using dataset GSE26554.
Camera FRY GAGE GSEA GSVA ORA PAGE PLAGE ROAST
FRY 0.00e+00
GAGE 0.00e+00 1.05e-128
GSEA 3.920e-67 0.00e+00 0.00e+00
GSVA 0.00e+00 3.78e-60 3.44e-24 0.00e+00
ORA 5.00e-05 0.00e+00 0.00e+00 5.33e-39 0.00e+00
PAGE 9.58e-105 0.00e+00 1.90e-320 9.41e-285 0.00e+00 2.07e-173
PLAGE 4.94e-324 0.00e+00 0.00e+00 0.00e+00 0.00e+00 0.00e+00 0.00e+00
ROAST 0.00e+00 1.76e-12 1.03e-65 0.00e+00 3.86e-65 0.00e+00 0.00e+00 0.00e+00
ssGSEA 0.00e+00 0.00e+00 0.00e+00 4.94e-324 0.00e+00 4.94e-324 0.00e+00 3.06e-19 0.00e+00
Note that a p-value of 0.00e+00 is smaller than the representable floating-point precision.
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
54
