
A General Framework for Exact Partially Local Alignments
Falco Kirchner
1
, Nancy Retzlaff
2,1
and Peter F. Stadler
1,2,3,4,5
1
Bioinformatics Group, Department of Computer Science, Interdisciplinary Center for Bioinformatics,
and Competence Center for Scalable Data Services and Solutions Dresden/Leipzig, Universit
¨
at Leipzig,
H
¨
artelstraße 16-18, D-04107 Leipzig, Germany
2
Max Planck Institute for Mathematics in the Sciences, Inselstraße 22, D-04103 Leipzig, Germany
3
Institute for Theoretical Chemistry, University of Vienna, W
¨
ahringerstraße 17, A-1090 Wien, Austria
4
Santa Fe Institute, 1399 Hyde Park Road, Santa Fe NM 87501, U.S.A.
Keywords:
Multiple Sequence Alignments, Dynamics Programming.
Abstract:
Multiple sequence alignments are a crucial intermediate step in a plethora of data analysis workflows in com-
putational biology. While multiple sequence alignments are usually constructed with the help of heuristic
approximations, exact pairwise alignments are readily computed by dynamic programming algorithms. In
the pairwise case, local, global, and semi-global alignments are distinguished, with key applications in pat-
tern discovery, gene comparison, and homology search, respectively. With increasing computing power, exact
alignments of triples and even quadruples of sequences have become feasible and recent applications e.g. in
the context of breakpoint discovery have shown that mixed local/global multiple alignments can be of practi-
cal interest. vaPLA is the first implementation of partially local multiple alignments of a few sequences and
provides convenient access to this family of specialized alignment algorithms.
1 INTRODUCTION
Global multiple alignments are typically constructed
as intermediate data structure to support a compara-
tive or evolutionary analysis homologous sequences.
Alignment problems are naturally treated as optimiza-
tion problems: a scoring function evaluates the sim-
ilarities in an alignment column and/or the pattern
of gaps. Multiple alignments are almost exclusively
treated globally, that is, all parts of the input sequence
is scored. The notion of “local multiple alignments”
appears mostly in the context of phylogenetic foot-
printing (Lukashin and Rosa, 1999; Blanchette et al.,
2002) and related pattern discovery problems (Tabei
and Asai, 2009), where substrings are considered that
appear with a limited number of mismatches in some
or all input sequences.
Local variants of sequence alignment, on the one
hand, play an important role in pairwise alignments.
Local alignments, i.e., maximally similar substrings
within pairs of longer sequences, are a natural way to
identify conserved domains. The semi-global variant
of pairwise alignment, in which one sequence, usu-
ally called “query”, is expected to appear as approxi-
mate substring of a larger “subject”, again is a natural
formalization of homology search, implemented e.g.
in gotohscan (Hertel et al., 2009). Overlap align-
ments (Jones and Pevzner, 2004) allowing free end
gaps on all sequences have applications e.g. in se-
quence assembly (Rausch et al., 2009). Until recently,
the generalization of these variants to more than two
sequences has received very little attention.
Pairwise alignment problems can be solved ex-
actly for a wide range of cost models by means of
dynamic programming. In fact, the algorithms of
Needleman and Wunsch (1970) for global alignments,
Smith and Waterman (1981) for local alignments, and
the extension to affine gap costs by Gotoh (1982) are
among the early, paradigmatic example of dynamic
programming. The basic recursive structure is read-
ily extended to more than two input sequences (Car-
rillo and Lipman, 1988; Lipman et al., 1989); the
time and space complexity, however, grows expo-
nentially with the number of sequences. Exact dy-
namic programming solutions thus have been used in
practice only for 3-way (Gotoh, 1986; Dewey, 2001;
Konagurthu et al., 2004; Kruspe and Stadler, 2007)
or 4-way (Steiner et al., 2011) alignments. Since
multiple sequence alignment problems (for arbitrary
numbers of input sequences X are typically NP-hard
194
Kirchner, F., Retzlaff, N. and Stadler, P.
A General Framework for Exact Partially Local Alignments.
DOI: 10.5220/0007380001940200
In Proceedings of the 12th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2019), pages 194-200
ISBN: 978-989-758-353-7
Copyright
c
2019 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
(Kececioglu, 1993; Wang and Jiang, 1994; Bonizzoni
and Della Vedova, 2001; Just, 2001; Manthey, 2003;
Elias, 2006), they are solved by heuristic approxima-
tion algorithms, see Chatzou et al. (2016), Baichoo
and Ouzounis (2017) or Nute et al. (2018) for a recent
reviews.
As the exact 3-way and 4-way alignments have in-
creased in usage, variants of the problem that com-
bine local and global alignments have been proposed
for specialized application scenarios. Al Arab et al.
(2017) considered the fate of sequences in the wake of
mitochondrial genome rearrangements by simultane-
ously comparing the rearranged region to both of its
ancestors. This approach made it possible to distin-
guish tandem duplication random loss (TDRL) from
reversal or transposition events. This specialized 3-
way alignment problem suggested the need to develop
a general theoretical framework for alignments that
consider part of their input local and part global. As
shown by Retzlaff and Stadler (2018) it is possible
– and convenient – to allow the user determine sep-
arately for each input sequence and each of its ends,
whether it is to be treated as global, i.e., deletions of
a prefix or a suffix are penalized, or as local, allow-
ing the omission of prefixes or suffixes at not cost.
We will briefly outline the theoretical results in the
following section. While the presentation by Retzlaff
and Stadler (2018) is purely theoretical and did not
supply a reference implementation, the present con-
tribution closes this gap.
2 THEORY
The basic idea behind the framework of Retzlaff and
Stadler (2018) boils down to two ingredients: (1)
Each input sequence is either local or global on the
left and either local or global on the right. This is
entirely the user’s choice and provided with the in-
put. (2) In a particular alignment column, a sequence
may be inactive (if up to this column its prefix is con-
sidered unaligned), active (if it contributes to the col-
umn either with one of its characters or with a gap
this is scored), or it is dead (if its suffix is consid-
ered unaligned). Consequently a left-local sequence
starts out inactive, while a left-global sequence starts
out active. Correspondingly, a right-global input is
still active at the end of the alignment, while a right-
local sequence must be dead at the end of the align-
ment. The partially local alignment problems can be
solved by dynamic programming just as the classic
pairwise problems mentioned in the introduction. As
usual, a scoring (memoization) table S holds the op-
timal alignments of prefixes. The only difference is
3
2
1
3
2
1
{1,2}|{}
{1,2,3}|{}
{1}|{2}
{1,3}|{2}
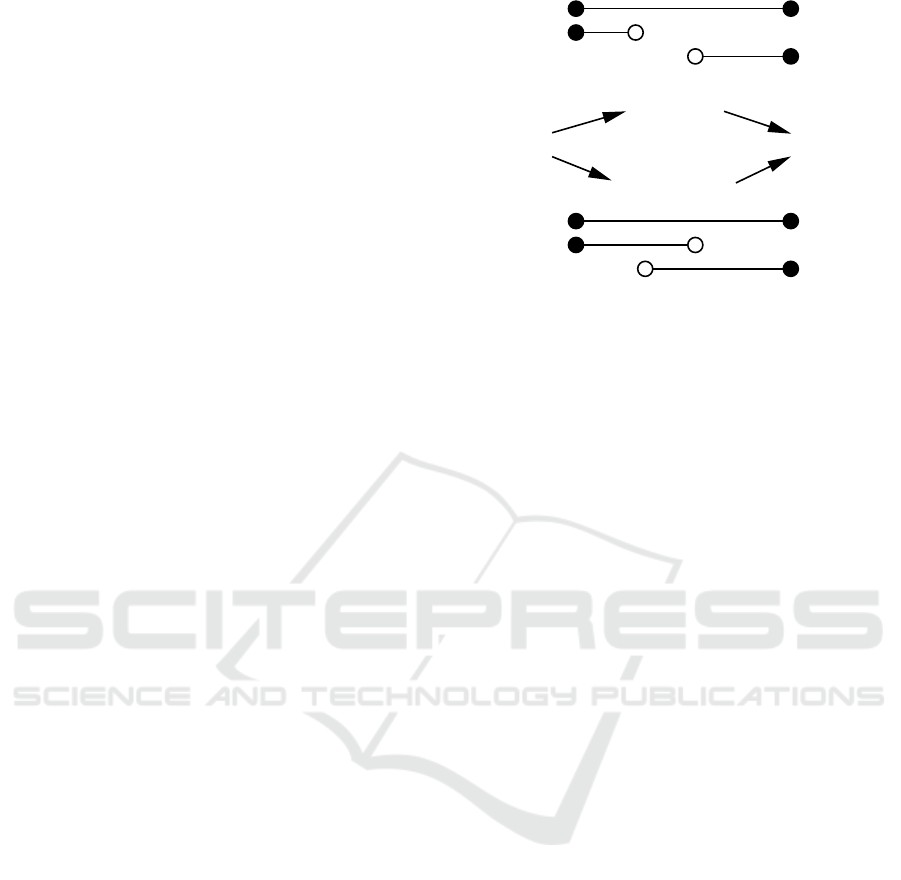
Figure 1: Schematic representation of the breakpoint align-
ment model of (Al Arab et al., 2017), with global reference
sequence 1, right-local prefix 2, and left-local suffix 3. The
initial and terminal states of valid alignments are therefore
A|D = {1,2}|{} and {1,3}|{2}, respectively. There are two
distinct path of transitioning between these states with in-
termediates states {1}|{2} (if 2 and 3 do not overlap) and
{1,2,3}|{} (if 2 and 3 overlap in their aligned part). The
black bullets indicate that the correspond end of the se-
quence is present in the alignment, open circles indicate the
prefixes of suffixes remain unaligned.
that S now depends not only on the length of prefixes
but also on the state (inactive, active, or dead) of each
sequence in a given column of the alignment. It is
sufficient to record the set A of active and D of dead
sequences, since the inactive sequences are given by
X \ {A ∪D}. As the alignment progresses, an inactive
sequence may become active only once, and an active
may transit at most once to the dead state. Consecu-
tive alignment columns thus have state pairs (A
0
,D
0
)
and (A,D) that are comparable w.r.t. the partial order
(A
0
,D
0
) (A, D) ⇐⇒
(
A
0
∪ D
0
⊆ A ∪ D
D
0
⊆ D
(1)
The state changes can be performed stepwisely. As
shown by Retzlaff and Stadler (2018), (A
0
,D
0
) is an
immediate predecessor of (A,D) if either exactly on
one inactive sequence become active or exactly one
active sequence transitions to the dead state. We
denote this relation by ≺≺. The initial condition is
A = A
0
, where A
0
is the set of left-global sequences,
and D =
/
0.
In a quite general form (which uses the heuristic
version of the affine gap cost model for more than two
sequences), the recursion for the optimal alignment
score are of the form
S
(A,D)
I
= max
max
π
h
S
(A,D)
I−π
+ s(π)
i
max
(A
0
,D
0
)≺≺(A,D)
h
S
(A,D)
I
+ s
∗
i
(2)
The variable π (a non-null binary vector) denotes gap
pattern in the last alignment column, the lower multi-
A General Framework for Exact Partially Local Alignments
195

index I describes the lengths of the prefixes included
in the alignment including this column. Thus I − π
is the vector of prefix lengths in the previous column.
The scoring function s( .) in the most general form
depends on both gap patterns as well as the actual se-
quence entries. The second alternative does not move
in the alignment but changes the state, a step that may
also be associated with a cost s
∗
, which in the most
general case may depend on I, π,A,D,A
0
,D
0
. Equa-
tion (2) describes the recursion for additive scores.
The special case that a sequence k that is both left-
and right-local remains completely unaligned is han-
dled by a directed transition from inactive to dead re-
stricted to I
k
= 0, see Retzlaff and Stadler (2018) for
details.
The notation is illustrated in Fig. 1 for the algo-
rithm introduced by Al Arab et al. (2017). In prin-
ciple, the recursions are easily extended to affine gap
costs. However, this incurs another factor (2
N
− 1)
in memory for N sequences since the scoring tables
S becomes explicitly dependent on the gap pattern of
the last column. The full recursions for the breakpoint
alignment model with affine gap costs, are given in
the appendix of Retzlaff and Stadler (2018).
The backtracing recursion is a rather straightfor-
ward generalization of the backtracing scheme for the
Smith-Waterman algorithm. The first step is to find
the optimal score of the partially local alignment. De-
note by (A
∗
,D
∗
) that unique maximal state w.r.t. ,
i.e., A
∗
is the set of all right-global sequences and D
∗
is the set of all right-local sequences. Hence A
∗
∪ D
∗
contains all input sequences. The optimal score is the
maximum over all multi-indices I with constraint that
I
k
= n
k
, the length of the k-th sequence, for all k ∈ A
∗
,
with the the maximum taken over all indices I
k
with
k ∈ D
∗
. The maximum value of I
k
determines the
right boundary of the right-local sequence. The recur-
sion then proceeds, as usual, to find the index or state
transition in Eq. (2). The backtracing recursion termi-
nates as soon as all left-global variables k ∈ A
0
have
reached the left end oft the sequences, i.e., I
k
= 0 for
all k ∈ A
0
. The left boundary of a left-local sequences
l /∈ A
0
equals the index I
l
at this point.
Equation (2) is the simplest way to explain the re-
cursive structure of the partially local alignment prob-
lem. It has the disadvantage that it provides more than
one way to obtain a particular partial alignment (char-
acterized by I,A,D) since state transition can be per-
formed in arbitrary order. As a consequence, Eq. (2)
cannot be used to compute partition functions over
alignments, and hence to obtain a probabilistic ver-
sion. As described in some detail by Retzlaff and
Stadler (2018), unambiguous recursions can be con-
structed by allowing state transitions from inactive
to active and from active to dead for a sequence k
only in conjunction with appending of a alignment
column for which π
k
= 1. At the same time, one
needs to consider also all possible state transitions
with (A
0
,D
0
) ≺ (A, D). That is,
S
(A,D)
I
= max
π
∗
max
(A
0
,D
0
)
h
S
(A
0
,D
0
)
I−π
+ s(π) +s
∗
i
(3)
where max
∗
(A
0
,D
0
)
runs over all (A
0
,D
0
) (A,D) such
that k ∈ A \ A
0
or k ∈ D \ D
0
implies π
k
= 1.
3 IMPLEMENTATION
We have implemented the two variants of the align-
ment algorithm for partially local alignments with ad-
ditive gap costs based on Eq. (2) and Eq. (3), respec-
tively. In the practical implementation of Eq. (3) we
omit the formal initial state (A
0
,
/
0) and separately ini-
tialize all left-local sequences k both in the inactive
and the active state for I
k
= 0. Similarly, we catch the
final states at the right end of right-local sequences
without explicitly considering a final transition to the
dead state after the last letter has been included into
the alignment. Eq. (3) in general requires the consid-
eration of more state changes in each step than Eq. (2).
While Eq. (2) only considers the Hasse diagram of the
partial order ≺, its transitive closure is required for
Eq. (3). On the other hand, Eq. (3) provides a very
convenient starting point for later extensions e.g. to a
probabilistic version.
vaPLA is written in Java and uses only standard
libraries. The source code is available on GitHub.
In principle, vaPLA is capable of accepting an arbi-
trary number of input sequences (in FASTA format)
together with information on whether each of their
ends is to be treated globally or locally. However,
the resource requirements quickly become prohibitive
with the number of input sequences and in particu-
lar the number of local ends. vaPLA first explicitly
constructs the Hasse diagram of the partial order ≺
for Eq. (3) and uses this information to allocated the
memoization tables for the recursions. The partial or-
der can be exported in .dot format and visualized us-
ing a standard graph drawing tools such as graphviz.
For Eq. (2) only the relation ≺≺, i.e., the edges of the
Hasse diagram, is used, while Eq. (3) makes use of
the entire partial order ≺.
The partial order determines the required re-
sources: one
∏
k∈A
O(n
k
)-size table is required for
each state (A,D). Writing g=|A
0
∩ A
∗
| for the num-
ber of global sequences, s = |A
0
\ A
∗
| + |A
∗
\ A
0
| for
the number sequences that are local and one end and
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
196
global at the other, and ` for the number of local se-
quences, there are
h = 1
g
2
s
3
`
(4)
distinct states, because global sequences are always
active, semi-local sequences change either state from
inactive to to active or from active to dead, while local
sequences can pass through all three states. All com-
binations of these states must be considered, since the
relative order (between sequences) of the state transi-
tions is not constrained. With one index variable iter-
ating over each of the N sequences of length O(n), the
memory requirements are O(n
N
) for each state. The
evaluation of the recursion requires O(2
N
) score com-
putations for each transition between columns and
states, resulting in an upper bound of O(2
N
n
N
h
2
) ef-
fort. The effort is reduced to O(2
N
n
N
hN) by imple-
menting Eq. (2) instead of Eq. (3) as shown in Fig. (3).
Both variants are available in the current implementa-
tion.
Backtracing is implemented in the usual manner:
starting from the position I
∗
and state (A
∗
,D
∗
) of the
optimal score, vaPLA computes the transition that re-
sulted in the optimal score. At present, co-optimal
solutions are not investigated. The first solution en-
countered is used. The procedure then continues iter-
atively until a valid start state is reached.
4 BENCHMARK
We use two well known benchmark protein databases
to test and evaluate vaPLA. OXBench (Raghava
et al., 2003) is a completely automatically generated
database whereas BAliBASE (Thompson et al., 2005)
has a manual step for cleaning initial alignments be-
fore the release. Both benchmark sets are intended for
global multiple alignments. We therefore inspected a
subset of the reference alignments and manually in-
spected overhanging ends, which we tagged for local
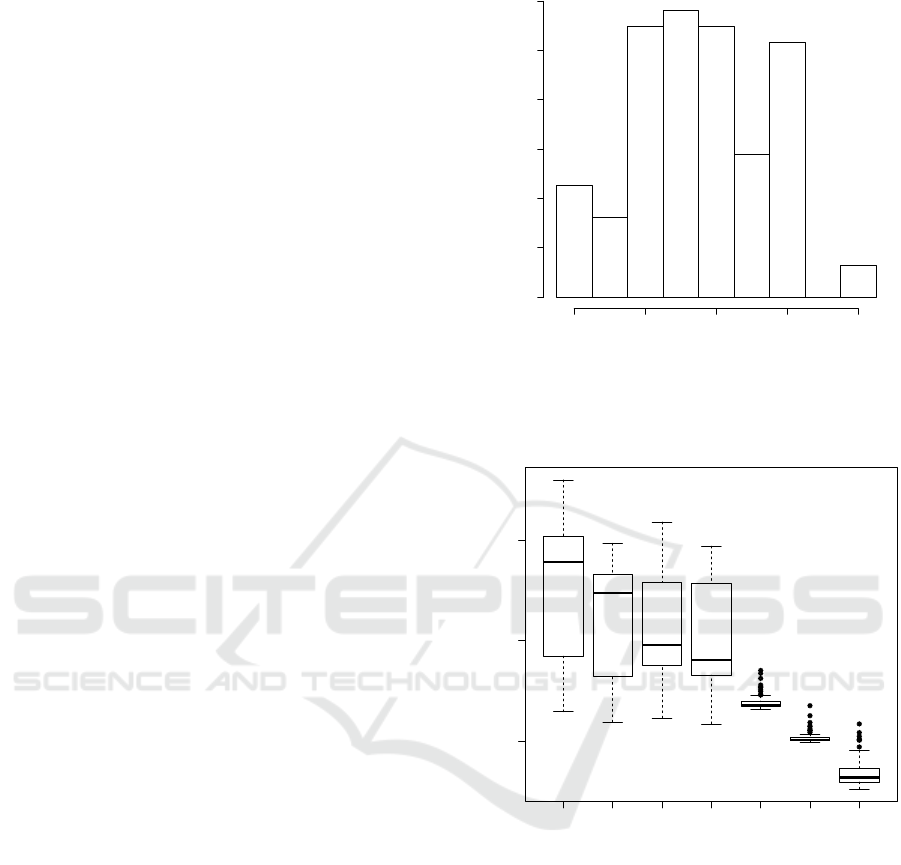
instead of global alignment. Figure 2 summarizes the
distribution of local ends in benchmark set used here.
These sequences are subsequently aligned with
vaPLA as well as three of the most common align-
ment tools: T-Coffee (Notredame et al., 2000; Chang
et al., 2014), MAFFT (Katoh et al., 2005; Nakamura
et al., 2018), and ClustalW (Larkin et al., 2007; Siev-
ers and Higgins, 2018). All three tools are based on
initial pairwise alignments and use essentially pro-
gressive schemes, hence presenting efficient heuris-
tics rather than exact solutions.
Comparing the performance for few and many lo-
cal ends, respectively, we can see in Fig. 3 that the
running time of vaPLA, as expected, strongly depends
#local ends in one alignment
frequency
0 2 4 6 8
0.020.00 0.01 0.03 0.04
0.05
0.06
Figure 2: Frequency distribution of the number of local ends
in the in the data set of benchmark alignments. Only a small
fraction of the alignments is global, while most test align-
ments have 2–6 local ends.
vaPLA>3
vaPLA*>3
vaPLA≤3
vaPLA*≤3
T-Coffee
MAFFT
ClustalW
5 10 15
log(t) [ms]
Figure 3: Running time of vaPLA for alignments with at
most three local ends (e.g. vaPLA≤ 3) and for alignments
with more than three local ends (e.g. vaPLA> 3) com-
pared to heuristic global aligners (T-Coffee, MAFFT, and
ClustalW) that are commonly used in large-scale bioin-
formatics applications. The label vaPLA refers to Eq. (2),
vaPLA* indicated the implementation following Eq. (3).
on how many local ends needs to be handled in one
alignment. Without local ends, i.e., for global align-
ments, the execution time of vaPLA is comparable
with T-Coffee. For partially local alignments we ob-
serve the expected exponential increase with the num-
ber of local ends. Since vaPLA is designed as a refer-
ence implementation of a much more expensive, ex-
act algorithm, it is clear that it cannot be competitive
in terms resource consumption. Figure 3 also com-
A General Framework for Exact Partially Local Alignments
197

vaPLA T−Coffee MAFFT ClustalW
50
60
70
80
90
100
Figure 4: Accuracy of vaPLA compared to the heuristic
global alignment tools T-Coffee, MAFFT, and ClustalW.
vaPLA T−Coffee MAFFT ClustalW
0
1
2
3
4
5
6
7
PSE
Figure 5: Position shift error of vaPLA compared to the
heuristic global aligners T-Coffee, MAFFT, and ClustalW.
pares the resource consumption for the implementa-
tion of Eqs. (2) and (3), resp. As expected, running
times decrease when Eq. (3) is used since one iterates
only over the immediate predecessors. The effect is
more pronounced for the data set with three or more
local ends, because their Hasse diagrams typically are
larger. Much more interesting than the comparisons
of running times, however, is the question whether
exact multi-way alignments yield an improvement in
accuracy.
The average accuracy, defined as AC = (L − f )/L
where L is the length of the alignment and f is
the number of columns deviating from the reference
alignment, is summarized in Fig. 4. The data show
that vaPLA provides a moderate but noticeable im-
provement relative to all three heuristics, although we
use a simple scoring model and none of the protein-
specific rules implemented e.g. in ClustalW.
The position shift error PSE as defined by
Raghava et al. (2003) serves as an alternative mea-
sure of alignment accuracy. Consider a pair of
(mis)matched positions i in sequence x and j in se-
quence y in the reference alignment. In the test align-
ment we consider the same position i in x and its
(mis)matched position j
0
in y and measure the dis-
tance δ = | j − j
0
|. A similar rule is used to compute
δ if there is an in/del between x and y at position i.
For the details we refer to (Raghava et al., 2003). The
PSE is the average of the contributions δ of all mis-
matched pairs in the reference alignment. Omitted
prefixes and suffixes at local ends do not enter the
PSE score. vaPLA exhibits significantly smaller po-
sition shift errors than the three heuristics, Fig. 5.
5 DISCUSSION
vaPLA is primarily intended as a reference implemen-
tation against which specialized partially local align-
ments can be tested and benchmarked. We anticipate
at least two use cases. First, vaPLA is useful to to
create test cases and help debugging during the devel-
opment phase of a specialized exact implementation.
More importantly, since vaPLA computed exact solu-
tions for a moderate number of input sequences, it can
be used to generate ground-truth data against which
faster heuristics can be compared. The current version
of vaPLA was not implemented to yield good perfor-
mance while we expect that substantial gains can be
achieved by parallelization with fine grained multi-
threading (Martins et al., 2001). Still it will need to be
tested whether such a solution is realizable in Java.
While biological sequences tend to be rather long
as compared to average word lengths we see a promis-
ing application to alignment of lexical items where
prefixing and suffixing seem to play even a bigger role
that in biology. Even though affixes can contain in-
formation, the root of words is most valuable when
doing cross-linguistics comparisons. For conceptual
examples we refer to (Retzlaff and Stadler, 2018).
The computational efforts for exact dynamic pro-
gramming algorithms often can be reduced exclud-
ing subsets of matches using bounds on the achiev-
able scores. Lossless filters for local pairwise align-
ments have been pioneered by Peterlongo et al. (2008,
2009). Ideas to prune the search space of the DP
problem are discussed e.g. by Schroedl (2005) or
Bilu et al. (2006). Quasi-alignments based on k-mer
matches can be employed as alignment anchors also
in a global context (Nagar and Hahsler, 2013). These
techniques may be used not only to reduce the com-
putational effort of the exact algorithm for input se-
quences of practical interest, but also might serve as
starting points for construcing efficient heuristics for
partially local MSA problems.
The framework of vaPLA lends itself to further ex-
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
198

tensions. First, it is easily possible to derive a prob-
abilistic version. This essentially entails a change in
the scoring from adding score contributions to mul-
tiplying with the corresponding Boltzmann factors.
The corresponding outside algorithm could easily be
constructed along the lines of H
¨
oner zu Siederdissen
et al. (2015). Another extension that could be realized
very easily is to enforce additional constraints on state
transitions. For example, it may be useful in a pattern-
based applications to allow the transition to and from
active only concurrently, i.e., at the same position rel-
ative to remaining input sequences.
Instead of treating state transitions in an acyclic
manner, it is also possible to allow multiple transi-
tions from active back to inactive. This would al-
low certain (context dependent) deletions to occur at
a unit cost. Such “exclusions” have rarely been con-
sidered in sequence alignment but are of some interest
for structured RNAs (Schirmer and Giegerich, 2013).
This idea may be of use in particular when sequences
are provided with structural annotation and deletions
of entire structural elements are to be scored in a spe-
cial way.
Advances in computing technology now make it
feasible to compute exact simultaneous solutions of
alignment problems with more than two sequences.
A combinatorial diversity of distinct alignment prob-
lems arises in this setting just by allowing to distin-
guish local and global ends separately for each input.
We suspect that additional variations on the theme
are of interest, e.g., requiring additional constraints
on pairwise overlaps. The framework and the imple-
mentation presented here is a first step towards an sys-
tematic exploration of this largely uncharted universe
of alignments, many of which we suspect will be of
practical use in computational biology.
ACKNOWLEDGMENTS
NR gratefully acknowledges the hospitality of the
Santa Fe Institute, where parts of this work were per-
formed, as well as travel support by the ASU-SFI
Center for Biosocial Complex Systems. The Compe-
tence Center for Scalable Data Services and Solutions
(ScaDS) Dresden/Leipzig is funded by BMBF grant
01IS14014B.
REFERENCES
Al Arab, M., Bernt, M., H
¨
oner zu Siederdissen, C., Tout,
K., and Stadler, P. F. (2017). Partially local three-way
alignments and the sequence signatures of mitochon-
drial genome rearrangements. Alg. Mol. Biol., 12:22.
Baichoo, S. and Ouzounis, C. A. (2017). Computational
complexity of algorithms for sequence comparison,
short-read assembly and genome alignment. Biosys-
tems, 156/157:72–85.
Bilu, Y., Agarwal, P. K., and Kolodny, R. (2006). Faster
algorithms for optimal multiple sequence alignment
based on pairwise comparisons. IEEE/ACM Trans.
Comp. Biol. Bioinf., 3:408–422.
Blanchette, M., Schwikowski, B., and Tompa, M. (2002).
Algorithms for phylogenetic footprinting. J Comput
Biol, 9:211–223.
Bonizzoni, P. and Della Vedova, G. (2001). The complexity
of multiple sequence alignment with SP-score that is
a metric. Theor. Comp. Sci., 259:63–79.
Carrillo, H. and Lipman, D. (1988). The multiple sequence
alignment problem in biology. SIAM J. Appl. Math.,
48:1073–1082.
Chang, J. M., Di Tommaso, P., and Notredame, C. (2014).
TCS: a new multiple sequence alignment reliability
measure to estimate alignment accuracy and improve
phylogenetic tree reconstruction. Mol. Biol. Evol.,
31:1625–1637.
Chatzou, M., Magis, C., Chang, J. M., Kemena, C., Bus-
sotti, G., Erb, I., and Notredame, C. (2016). Multiple
sequence alignment modeling: methods and applica-
tions. Brief Bioinform., 17:1009–1023.
Dewey, T. G. (2001). A sequence alignment algorithm with
an arbitrary gap penalty function. J. Comp. Biol.,
8:177–190.
Elias, I. (2006). Settling the intractability of multiple align-
ment. J. Comp. Biol., 13:1323–1339.
Gotoh, O. (1982). An improved algorithm for matching
biological sequences. J. Mol. Biol., 162:705–708.
Gotoh, O. (1986). Alignment of three biological sequences
with an efficient traceback procedure. J. theor. Biol.,
121:327–337.
Hertel, J., de Jong, D., Marz, M., Rose, D., Tafer, H.,
Tanzer, A., Schierwater, B., and Stadler, P. F. (2009).
Non-coding RNA annotation of the genome of Tri-
choplax adhaerens. Nucleic Acids Res., 37:1602–
1615.
H
¨
oner zu Siederdissen, C., Prohaska, S. J., and Stadler, P. F.
(2015). Algebraic dynamic programming over general
data structures. BMC Bioinformatics, 16:19:S2.
Jones, N. C. and Pevzner, P. A. (2004). An Introduction to
Bioinformatics. MIT Press, Cambride, MA. Problem
6.22.
Just, W. (2001). Computational complexity of multiple
sequence alignment with SP-score. J. Comp. Biol.,
8:615–623.
Katoh, K., Kuma, K.-i., Toh, H., and Miyata, T. (2005).
MAFFT version 5: improvement in accuracy of mul-
tiple sequence alignment. Nucleic Acids Res., 33:511–
518.
Kececioglu, J. D. (1993). The maximum weight trace prob-
lem in multiple sequence alignment. In Proceedings of
A General Framework for Exact Partially Local Alignments
199

the 4th Symposium on Combinatorial Pattern Match-
ing, volume 684 of Lecture Notes Comp. Sci., pages
106–119, Berlin. Springer.
Konagurthu, A. S., Whisstock, J., and Stuckey, P. J. (2004).
Progressive multiple alignment using sequence triplet
optimization and three-residue exchange costs. J.
Bioinf. and Comp. Biol., 2:719–745.
Kruspe, M. and Stadler, P. F. (2007). Progressive multiple
sequence alignments from triplets. BMC Bioinformat-
ics, 8:254.
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R.,
McGettigan, P. A., McWilliam, H., Valentin, F., Wal-
lace, I. M., Wilm, A., Lopez, R., Thompson, J. D.,
Gibson, T. J., and Higgins, D. G. (2007). Clustal W
and Clustal X version 2.0. Bioinformatics, 23:2947–
2948.
Lipman, D. J., Altschul, S. F., and Kececioglu, J. D. (1989).
A tool for multiple sequence alignment. Proc. Natl.
Acad. Sci. USA, 86:4412–4415.
Lukashin, A. V. and Rosa, J. J. (1999). Local multiple se-
quence alignment using dead-end elimination. Bioin-
formatics, 15:947–953.
Manthey, B. (2003). Non-approximability of weighted
multiple sequence alignment. Theor. Comp. Sci.,
296:179–192.
Martins, W. S., Del Cuvillo, J. B., Useche, F. J., and
Theobald, K. B.and Gao, G. R. (2001). A multi-
threaded parallel implementation of a dynamic pro-
gramming algorithm for sequence comparison. In Alt-
man, R. B. A., Dunker, A. K., Hunker, L., Lauderdale,
K., and Klein, T. E., editors, Pacific Symposium on
Biocomputing, volume 6, pages 311–322, Singapore.
World Scientific.
Nagar, A. and Hahsler, M. (2013). Fast discovery and visu-
alization of conserved regions in DNA sequences us-
ing quasi-alignment. BMC Bioinformatics, 14 (Suppl
11):S2.
Nakamura, T., Yamada, K. D., Tomii, K., and Katoh, K.
(2018). Parallelization of MAFFT for large-scale mul-
tiple sequence alignments. Bioinformatics, 34:2490–
2492.
Needleman, S. B. and Wunsch, C. D. (1970). A general
method applicable to the search for similarities in the
amino acid sequence of two proteins. J Mol Biol,
48:443–453.
Notredame, C., Higgins, D. G., and Heringa, J. (2000). T-
coffee: a novel method for fast and accurate multiple
sequence alignment. Journal of molecular biology,
302:205–217.
Nute, M., Saleh, E., and Warnow, T. (2018). Evaluating
statistical multiple sequence alignment in comparison
to other alignment methods on protein data sets. Syst
Biol.
Peterlongo, P., Pisanti, N., Boyer, F., Pereira do Lago, A.,
and Sagot, M.-F. (2008). Lossless filter for multiple
repetitions with Hamming distance. J. Discrete Algo-
rithms, 6:497–509.
Peterlongo, P., Sacomoto, G. A. T., Pereira do Lago, A.,
Pisanti, N., and Sagot, M.-F. (2009). Lossless filter
for multiple repeats with bounded edit distance. Alg.
Mol. Biol., 4:3.
Raghava, G. P. S., Searle, S. M. J., Audley, P. C., Bar-
ber, J. D., and Barton, G. J. (2003). OXBench:
a benchmark for evaluation of protein multiple se-
quence alignment accuracy. BMC Bioinformatics,
4:47.
Rausch, T., Koren, S., Denisov, G., Weese, D., Emde, A.-
K., D
¨
oring, A., and Reinert, K. (2009). A consistency-
based consensus algorithm for de novo and reference-
guided sequence assembly of short reads. Bioinfor-
matics, 25:1118–1124.
Retzlaff, N. and Stadler, P. F. (2018). Partially local multi-
way alignments. Math. Comp. Sci., 12:207–234.
Schirmer, S. and Giegerich, R. (2013). Forest alignment
with affine gaps and anchors, applied in RNA struc-
ture comparison. Theor. Comp. Sci., 483:51–67.
Schroedl, S. (2005). An improved search algorithm for opti-
mal multiple-sequence alignment. J. Artif. Intel. Res.,
23:587–623.
Sievers, F. and Higgins, D. G. (2018). Clustal Omega
for making accurate alignments of many protein se-
quences. Protein Sci., 27:135–145.
Smith, T. F. and Waterman, M. S. (1981). Identification
of common molecular subsequences. J Mol Biol,
147:195–197.
Steiner, L., Stadler, P. F., and Cysouw, M. (2011). A
pipeline for computational historical linguistics. Lan-
guage Dynamics & Change, 1:89–127.
Tabei, Y. and Asai, K. (2009). A local multiple alignment
method for detection of non-coding RNA sequences.
Bioinformatics, 25:1498–1505.
Thompson, J. D., Koehl, P., Ripp, R., and Poch, O. (2005).
BAliBASE 3.0: latest developments of the multiple
sequence alignment benchmark. Proteins: Structure,
Function, and Bioinformatics, 61:127–136.
Wang, L. and Jiang, T. (1994). On the complexity of multi-
ple sequence alignment. J Comput Biol, 1:337–348.
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
200
