
On-line Searching in IUPAC Nucleotide Sequences
Petr Proch
´
azka and Jan Holub
Department of Theoretical Computer Science, Faculty of Information Technology,
Czech Technical University, Th
´
akurova 2700/9, Prague 6, Czech Republic
Keywords:
Consensus Nucleotide Sequences, Genomic Sequences, Degenerate Pattern Matching, q-gram Inverted Index.
Abstract:
We propose a novel pattern matching algorithm for consensus nucleotide sequences over IUPAC alphabet,
called BADPM (Byte-Aligned Degenerate Pattern Matching). The consensus nucleotide sequences represent
a consensus obtained by sequencing a population of the same species and they are considered as so-called
degenerate strings. BADPM works at the level of single bytes and it achieves sublinear search time on average.
The algorithm is based on tabulating all possible factors of the searched pattern. It needs O (m + mα
2
logm)-
space data structure and O(mα
2
) time for preprocessing where m is a length of the pattern and α represents
a maximum number of variants implied from a 4-gram over IUPAC alphabet. The worst-case locate time is
bounded by O(nm
2
α
4
) for BADPM where n is the length of the input text. However, the experiments performed
on real genomic data proved the sublinear search time. BADPM can easily cooperate with the block q-gram
inverted index and so achieve still better locate time. We implemented two other pattern matching algorithms
for IUPAC nucleotide sequences as a baseline: Boyer-Moore-Horspool (BMH) and Parallel Naive Search (PNS).
Especially PNS proves its efficiency insensitive to the length of the searched pattern m. BADPM proved its strong
superiority for searching middle and long patterns.
1 INTRODUCTION
DNA sequencing is nowadays the integral part of sev-
eral disciplines like personalized medicine, human
genetics and oncology, forensic biology, microbiol-
ogy and virology. The demand for cheap sequenc-
ing induced the evolution of High-Throughput Se-
quencing (HTS) technologies that can sequence large
stretches of DNA in a massively parallel fashion and
that produce millions of DNA sequences simultane-
ously. The public sources report the necessary time
per one run in the order of hours and the cost per
one million bases lower than 0.012 USD
1
which is
1121 USD per human genome. Thanks to the general
availability of the sequencing, we face the challenge
of analyzing rapidly increasing number of genomic
sequences, which includes their effective storage in
the form allowing extremely fast searching.
The accessibility of sequencing methods has
also enabled the development of projects focused
on sequencing the population of many individuals
of the same species. These projects include the
1000 Genomes Projects (Consortium, 2011) and the
UK10K project (Consortium, 2015). Each sequenced
1
https://www.genome.gov/sequencingcostsdata/
population uses its ‘reference genome’, which forms
the basis for studying genetic variations for other in-
dividuals. Pan-genomics (Marschall, 2018) shifts the
reference genome to a representation of all genomic
content in a certain species or a phylogenetic clade.
A consensus sequence drawn from an entire popula-
tion is one of the forms that the reference genome can
take. The consensus sequence can be expressed as a
degenerate string over a degenerate alphabet. We give
a simple example of a consensus sequence expressed
in IUPAC alphabet
2
in Figure 1. The consensus repre-
sents an alignment of seven different organisms. The
different bases are emphasized with gray color. The
consensus base is given in blue color and it means that
more than one solid bases are valid (e.g. for R both A
or G symbols are valid).
Knuth-Morris-Pratt (KMP) (Knuth et al., 1977)
is one of the most famous pattern matching algo-
rithms and the first one ensuring the worst-case time
linear with the length of the text T . Boyer-Moore
(BM) (Boyer and Moore, 1977) family algorithms
represent backward pattern matching approach. BM
algorithm allows skipping of some characters which
leads to lower than linear average time. There exist
2
https://iupac.org
66
Procházka, P. and Holub, J.
On-line Searching in IUPAC Nucleotide Sequences.
DOI: 10.5220/0007382900660077
In Proceedings of the 12th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2019), pages 66-77
ISBN: 978-989-758-353-7
Copyright
c
2019 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

T C C A G C G C T T A C T C T A T A C C T A A
T C C A G C A C T T A C T C T G T G C C C G C
T C C A G C A C T T A C T C T G T G C C C A C
T C C A G C A C T T A C T C T G T G C C C A C
T C C A G C A C T T A C T C T G T G C C C G C
T C T A G C A C T T A C T C T A T G C C T G C
T C T A G C A C T T A C T C T A T G C C T G C
homo sapiens:
pan paniscus:
chlorocebus sabaeus:
macaca fascicularis:
macaca mulatta:
papio anubis:
callithrix jacchus:
CONSENSUS:
T C Y A G C R C T T A C T C T R T R C C Y R M
Figure 1: Consensus sequence over IUPAC alphabet for dif-
ferent species (chromosome 7: 55 187 593 - 55187 615).
also other variations of this algorithm given by Hor-
spool (Horspool, 1980) or Sunday (Sunday, 1990).
Suffix automaton (often called DAWG – Determin-
istic Acyclic Word Graph) is the essence of another
algorithm achieving sublinear average time BDM
(Backward DAWG Match) (Crochemore and Rytter,
1994). The suffix automaton of the reversed pattern
performs backward searching for the pattern. The
byproduct of the search is always the longest prefix
of the pattern occurring at that position in the text
which ensures safe shifting for BDM. Another ap-
proach is to use non-deterministic instead of deter-
ministic automata for searching in the text. So-called
bit-parallelism (D
¨
om
¨
olki, 1964; Baeza-yates, 1992)
proved to be a very simple way to simulate the non-
deterministic automaton. It exploits the parallelism
provided by bitwise operations in terms of one com-
puter word. It can accelerate the operations up to a
factor w, where w is the number of bits in the com-
puter word. Bit-parallelism is particularly efficient
for the patterns with size lower than the size of the
computer word m ≤ w. Navarro et al. applied the
bit-parallelism to simulate the suffix automaton and
they proposed BNDM algorithm (Navarro and Raf-
finot, 1998) that achieved 20%-25% improvement in
search time in comparison to its deterministic version
BDM.
The standard pattern matching was naturally ex-
tended to the problem of pattern matching in de-
generate strings, i.e. the strings where each degen-
erate symbol can represent a subset of solid sym-
bols. The very first practical algorithm addressing
the pattern matching in degenerate strings was agrep
utility (Wu and Manber, 1992). Next, Navarro et
al. (Navarro and Raffinot, 1998) introduced an exten-
sion of BNDM NR-grep (Navarro and Raffinot, 2002)
allowing classes of characters at each position of the
searched pattern. Holub et al. (Holub et al., 2008)
implemented a version of Boyer-Moore-Sunday algo-
rithm (Sunday, 1990) for degenerate strings and ex-
perimentally proved its superiority over BNDM for
standard pattern lengths. Iliopoulos et al. (Iliopou-
los et al., 2008) presented their algorithm based on
transformation into the ‘restricted pattern matching
problem’. Their implementation runs in O(n) time
where n is a size of the input text for sufficiently
small patterns and DNA/RNA alphabet. The algo-
rithm based on Landau and Vishkin’s algorithm was
proposed by Crochemore (Crochemore et al., 2015).
They achieved interesting upper bound O(kn), where
k is maximum number of degenerate positions in the
text and n is the length of the text. However, the
experimentally evaluated implementation of the algo-
rithm was not provided. Finally, the concept of ‘Elas-
tic Degenerate Strings’ was introduced in (Iliopou-
los et al., 2017). Elastic Degenerate Strings provide
another alternative to represent the consensus/pan-
genomic sequences. Later, the practical implementa-
tion of the pattern matching algorithm in Elastic De-
generate Strings was given in (Grossi et al., 2017)
and a version of algorithm allowing errors was pre-
sented in (Bernardini et al., 2017). Recently, Cisłak et
al. (Cisłak et al., 2018) presented SOPanG algorithm
solving elastic degenerate string matching problem in
the same upper bound O(Nd
m
w
e), however, achieving
an order of magnitude better time than (Grossi et al.,
2017) in practical experiments.
The nucleotide consensus sequences generated
from vcf (Variant Call Format) files have the follow-
ing properties. They have very limited number of dis-
tinct symbols in the alphabet (16 symbols of IUPAC
alphabet of which only 4 symbols are solid). At the
same time, the sequences prove a very low rate of de-
generate symbols (between two and three percent for
all chromosomes). The two aforementioned proper-
ties led us to the idea to store the consensus sequences
as a simply encoded strings (over the alphabet of only
solid symbols) together with marking the relatively
rare degenerate positions. Our algorithm BADPM is a
natural extension (for degenerate strings) of the BAPM
algorithm presented in (Proch
´
azka and Holub, 2017).
It is based on tabulating all possible factors of the
searched pattern. The algorithm performs searching
for all pattern factors in its basic loop. After every
factor occurrence, the occurrence of the whole pattern
must be confirmed.
BADPM works at the level of bytes which ensures
its high speed. This implies that the length of the
tabulated factors is expected to be a multiple of 4.
Our experiments proved that the factor length 8 sym-
bols is ideal trade-off between factor occurrence fre-
quency on one side and memory consumption and the
length of the shift on the other side. However, this ap-
proach ensures sublinear time complexity on average
since frequent shifting in the text can be applied. This
holds especially for longer patterns since the shift is
implied from the pattern length. The space for a sim-
ple data structure (storing the encoded consensus se-
quence) is upper bounded by O(nα + n logn) where n
is a size of the text and α is a maximum possible vari-
ants provided by 4-gram of IUPAC degenerate sym-
On-line Searching in IUPAC Nucleotide Sequences
67

bols. In practice, the needed space is close to O(n)
since the rate of degenerate symbols in the consensus
sequences is low and the number of possible variants
provided by the occurring 4-grams is low as well. The
ensured worst-case search time is O(nm
2
α
4
) where m
is a size of the searched pattern. However, our ex-
periments proved that the factor hit rate is very low.
Thus, the real time complexity is linear for short pat-
terns (16 ≤ m ≤ 32) and sublinear for longer patterns
(m > 32).
The rest of the paper is organized as follows. We
give definitions of some basic notions in Section 2.
The Section 3 is dedicated to definition and detailed
description of BADPM algorithm. We define PNS (Par-
allel Naive Search) algorithm as a baseline for experi-
mental comparison with BADPM in Section 4. The Sec-
tion 5 summarizes experimental results performed on
real genomic data. We give the conclusion and some
ideas for future work in Section 6.
2 BASIC NOTIONS
Let x = x
1
x
2
..x
n
be a string composed of single sym-
bols x
i
of a finite ordered alphabet Σ. The length of
the string x is n = |x|. The size of the alphabet Σ is
σ = |Σ| = O(1). The start position i and the length
j define so-called factor (or substring) denoted by
x
i, j
= x
i
..x
i+ j−1
. A factor with i = 0 is called prefix
and a factor with i + j − 1 = n is called suffix of the
string x. We denote by ε so-called empty string of
length 0. The problem of string pattern matching is
to find all occurrences of a pattern P = p
1
p
2
..p
m
in a
text T = t
1
t
2
..t
n
where both strings are composed of
symbols from the same alphabet Σ and m n. Partic-
ularly, we can distinguish two tasks: (i) count when
number of occurrences of P in T is reported and (ii)
locate when exact positions of the occurrences of P
in T are reported.
Pattern substitution method (Manber, 1997) is a
compression method when q-grams of symbols of the
input text T (i.e., Σ
q
) are substituted with an assigned
byte value b where b ∈ {0,1, ...,255}. The pattern
matching in the compressed (encoded) text means to
find all occurrences of the compressed pattern P
C
in
the compressed text T
C
(both defined over the alpha-
bet of byte values b ∈ {0,1,...,255}).
A symbol that represents only a single value is
called solid symbol. For DNA nucleotide sequences,
we consider the following alphabet of solid sym-
bols Σ = {A,C, G,T }. Degenerate symbol is called
a symbol that represents a subset of solid sym-
bols. The number of possible degenerate symbols
is limited by 2
|Σ|
− 1. The degenerate symbol can
be represented as a union of covered solid sym-
bols (e.g. [AC]) or as a specific single symbol (e.g.
M). For DNA consensus sequences, we consider the
following alphabet of degenerate symbols Σ
IUPAC
=
{A,C,G,T,R,Y, S,W,K,M, B,D, H,V,N}. The map-
ping IUPAC symbols to the solid symbols is given in
Table 1.
Table 1: Mapping IUPAC degenerate symbols into the sub-
sets of solid symbols.
IUPAC symbol
Subset Bit coding
A {A} h0001i
C {C} h0010i
G {G} h0100i
T {T } h1000i
R {A,G} h0101i
Y {C,T } h1010i
S {C,G} h0110i
W {A,T } h1001i
K {G,T } h1100i
M {A,C} h0011i
B {C,G,T } h1110i
D {A,G, T} h1101i
H {A,C, T} h1011i
V {A,C,G} h0111i
N {A,C,G, T} h1111i
For the degenerate text and the degenerate pattern
we can distinguish the following three pattern match-
ing problems (Iliopoulos et al., 2008). We design our
algorithm to solve the last problem which implies that
it can solve also the previous two problems.
Problem 1. Given a degenerate text T and a pattern
P . The problem is to find all the occurrences of P in
T i.e. to find all i such that for all j in [1,m], P
j
∈
T
i+ j−1
.
Problem 2. Given a text T and a degenerate pat-
tern P . The problem is to find all the occurrences
of P in T i.e. to find all i such that for all j in [1,m],
T
i+ j−1
∈ P
j
.
Problem 3. Given a degenerate text T and a degen-
erate pattern P . The problem is to find all the occur-
rences of P in T i.e. to find all i such that for all j in
[1,m], T
i+ j−1
∩ P
j
6=
/
0.
Traditional inverted index consists of two major
components: a vocabulary storing all distinct words
occurring in the text T and a set of posting lists stor-
ing positions of all occurrences of a given word in
the text T . The vocabulary of a q-gram inverted in-
dex (Puglisi et al., 2006) is composed of all possible
q-grams of the alphabet Σ, i.e., Σ
q
. For the purpose
of block indexing we split the indexed text into single
blocks of a defined fixed size. The posting lists of a
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
68

block inverted index then store addresses of the blocks
covering the exact positions of occurrences. The exact
positions are determined in the next step when a stan-
dard pattern matching method is performed in terms
of the preselected blocks.
In later description of the algorithm, we use C-
like syntax for bitwise operations. Particularly, we
use | for bitwise-or, & for bitwise-and, for shift-
left operation and for shift-right operation.
3 BYTE-ALIGNED DEGENERATE
PATTERN MATCHING
BADPM (Byte-Aligned Degenerate Pattern Matching)
algorithm is focused on searching in consensus nu-
cleotide sequences. It exploits their two basic proper-
ties: (i) very low size of the alphabet, (ii) very low fre-
quency of degenerate symbols in the text. Consider-
ing the alphabet expressing the values of solid bases,
the alphabet has only four symbols. Considering even
the degenerate bases, we still end up with fifteen sym-
bols at most. Furthermore, we experimentally tested
vcf files from the 1000 Genomes Projects (Consor-
tium, 2011) and we found that the frequency of the
degenerate symbols varies from 2 to 3 percent for dif-
ferent chromosomes.
The idea behind BADPM is to consider the consen-
sus (degenerate) sequence as a solid string and to en-
code it using a simple substitution encoding (Manber,
1997). The encoding method is defined as: f : Σ
4
7→ B
where B = {0, 1,. .. ,255} and b ∈ B represents a byte
value that is composed as a concatenation of bit cou-
ples given by the single symbols of the 4-gram s ∈ Σ
4
(A → 00, C → 01, G → 10, T → 11). The degen-
erate symbols in the string are explicitly marked us-
ing variantPos array. Two other auxiliary arrays are
needed to correctly evaluate the degenerate symbols
in the string. Array variantNum stores the number
of byte variants (implied from all degenerate sym-
bols of the byte). Array variants stores the byte vari-
ants/values implied from the degenerate symbols oc-
curring in the given byte. The solid symbols of the
byte are kept as a left and/or right context in terms
of the byte. This approach implies a space waste be-
cause of storing the solid symbols in the byte as the
contexts, however, it accelerates the search since the
byte operation level is kept. The aforementioned aux-
iliary arrays are very easy to serialize. Together with
the base sequence, between 30 and 35 percent of the
original size is used to store the BADPM encoded text.
On the other hand, the encoded IUPAC sequence (us-
ing a simple substitution encoding - 2 IUPAC symbols
encoded with one byte) requires exactly 50 percent of
the original size.
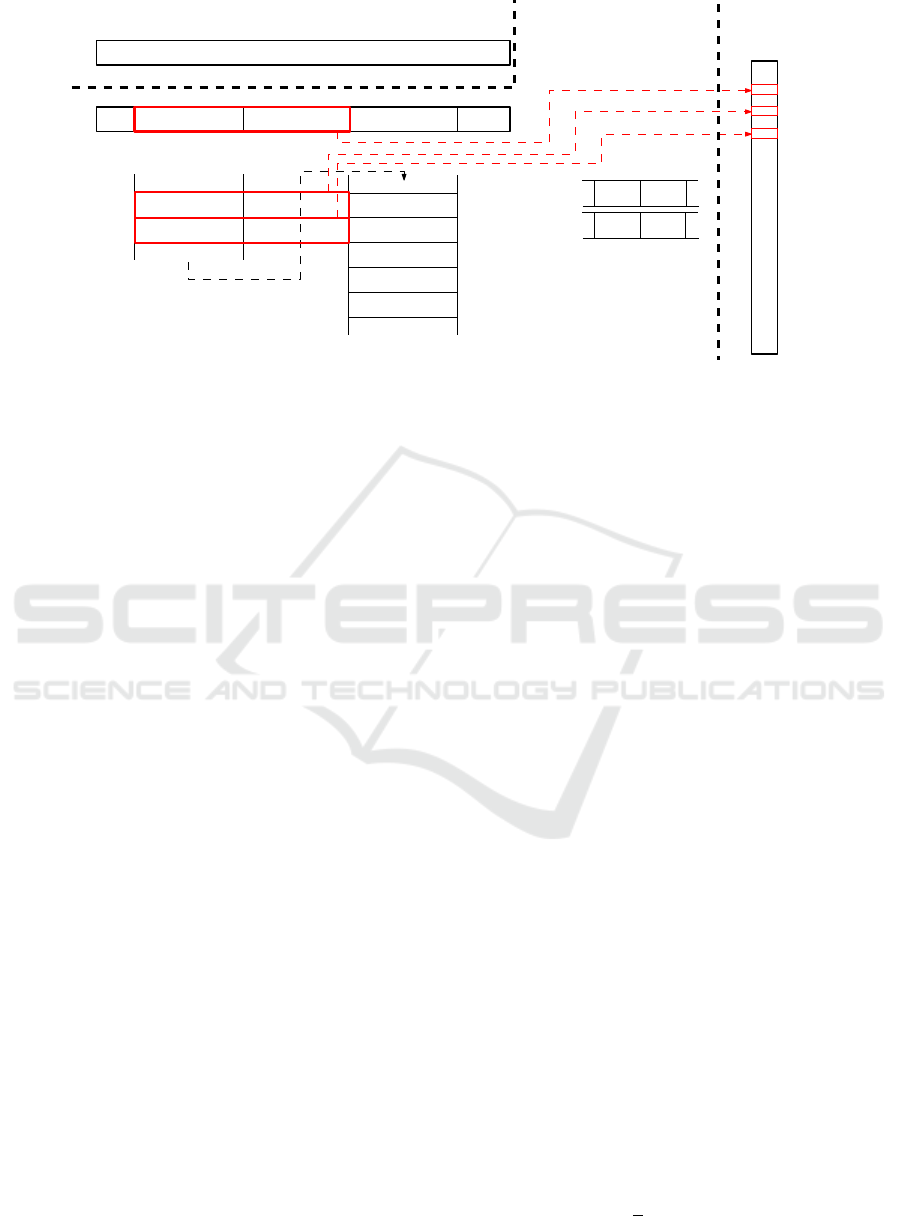
The BADPM encoded text is depicted in Figure 3.
Let α denotes a maximum number of variants implied
from a 4-gram over IUPAC alphabet and n denotes
the length of the input text. The 4-gram over IUPAC
alphabet providing most variants is clearly hNNNNi
that provides α = 4
4
= 256 variants. We state the fol-
lowing space complexity of the encoded sequence in
bits. Array variantNum storing the number of byte
variants implied from the corresponding degenerate
position clearly requires O(nlog α) space at most. At
most O(nlog n) space is required for variantPos ar-
ray. At most O(nα) space is needed for variants ar-
ray storing all byte variants. The encoded base se-
quence intuitively needs O(n) space. Thus, the total
space complexity is limited by O(nα + n logn). How-
ever, the real space needed for encoded consensus se-
quences fits between 30 and 35 percent of their origi-
nal/unencoded size.
The following algorithms and their complexities
correspond to BADPM variant optimized for Problem 3
if not stated others. BADPM needs a simple prepro-
cessing of the pattern that includes tabulating of all
pattern factors of a given length. The optimal length
of the factors is 8 symbols which means two-byte-
long factors of the encoded pattern. Figure 2 depicts
a simple data structure used to store the tabulated en-
coded factors of the pattern and it demonstrates also
BADPM preprocessing phase when this data structure
is filled. The dictionary data structure is depicted in
the right part of the figure and its main part is an ar-
ray (denoted as dictionary) with 65536 entries (corre-
sponding to 65536 different two-byte values). Every
entry can contain a pointer to a list which stores all
occurrences of the factor (corresponding to the entry)
in terms of the pattern. Each element of the list is a
couple (offset, alignment). The offset o represents a
byte position of the factor in the encoded pattern and
it is easy deducible from its starting position i in the
raw/unencoded pattern o = b
i−1
4
c. The alignment a
represents a position of the factor in terms of the byte
and it can be computed as a = (i − 1) mod 4. We
state the following space complexity of the dictionary
(preprocessed pattern) in bits. The offset o requires
O(log
m
4
) bits, however, the alignment a needs only
a constant number of bits O(1). Thus, one element
of the list requires O(logm) space. The space of the
dictionary array is O(α
2
) and every list can possibly
contains up to m − 7 elements. This implies the total
space complexity O(mα
2
logm).
In practice, the algorithm uses byte coding for
simpler and faster memory operations. Suppose the
length of the raw pattern m = 128 bases which im-
plies 32 bytes for the encoded pattern P
C
. Further-
On-line Searching in IUPAC Nucleotide Sequences
69

... C
00 01 10 11 00 00 11 11 00 00 11...
... 01 11 11 11 00 11 11 11 00 00 01
A C
G
T
A
A T ...
T
T
A A C
T
alignment = 0
alignment = 1
alignment = 2
alignment = 3
0
6 927
27 708
32 575
50 115
53 185
62 448
64 764
45 296
dictionary
offset
alignment
n
B
− 2
1
A → 00
C → 01
G → 10
T → 11
0
1
0
0
0
2
3
0
n
B
− 1
0
n
B
− 2
3
n
B
− 2
2
...
...
...
...
...
...
...
...
Preprocessing process Preprocessed pattern
T
A
A T
T
T
A
T
A C
G
T
A
A
T
...
T
A
A T
A C
G
T
A
A T ...
T
A
A T
A C
G
T
A
A T ...
T
A
A T
00 01 10 11 00 00 11 11 00 00 11...
00 01 10 11 00 00 11 11 00 00 11...
00 01 10 11 00 00 11 11 00 00 11...
... C
... 01 11 11 11 00 11 11 11 00 00 01
T
T
A
A C
T
T
T
A
T
... C
... 01 11 11 11 00 11 11 11 00 00 01
T
T
A
A C
T
T
T
A
T
... C
... 01 11 11 11 00 11 11 11 00 00 01
T
T
A
A
C
T
T
T
A
T
Figure 2: BADPM preprocessing phase for different byte alignments.
more, suppose every 16
th
symbol is degenerate imply-
ing 3 byte variants on average. BADPM needs to store
128−7+
128
16
×2 = 137 factors (elements of the lists).
Suppose a simple byte code used to store the offset o
and the alignment a for every factor. Only two bytes
are consumed for every pair (o,a) and still all the in-
formation is encoded at the level of bytes. Thus, for
BADPM the total space is 137 × 2 + 65 536 = 65 810
bytes, plus an overhead needed to implement the lists.
Still, the data structure easily fits into 65 kiB of mem-
ory and it can be kept in a fast level of the computer
memory.
Algorithm 1 describes preprocessing and search-
ing phase of BADPM. The function ENCODE is called
in the preprocessing phase. The function performs
the simple substitution encoding described above. Its
parameters are: the text to be encoded; the starting
index for encoding; and the number of bases/symbols
that need to be encoded. The function returns desired
encoded factor of the text. The function BUILDDIC-
TIONARY is responsible for constituting dictionary D
and storing the shifted versions of the pattern in the
array B. The while cycle (line 4) iterates over all
possible alignments a ∈ {0,1,2,3}. For every align-
ment, the number of bases/symbols that constitute the
longest byte sequence starting at i is computed (line 5)
and the corresponding encoded pattern is obtained
(line 6). The encoded pattern is stored for the given
alignment (line 7) and later is used for direct compar-
ison of bytes (the encoded text with the encoded pat-
tern). Next while cycle (line 9) iterates over all byte
couples of the encoded aligned pattern and all corre-
sponding variants (line 10) and it ensures storing the
couples (offset, alignment) to their corresponding lists
(line 13). E
j,2
represents a variant (composed of en-
coded solid symbols) provided by two-byte substring
of the encoded pattern E starting at position j.
The function BUILDMASK is another part of the
preprocessing. It generates all necessary masks pos-
sibly needed in the last step of the comparison (a pre-
fix and/or a suffix of the encoded pattern with the
corresponding part of the encoded text). Since the
prefix and the suffix are smaller than one byte the
masks are necessary to minimize the bitwise opera-
tions and therefore also the needed time. The function
stores the masks in single variables. The variable pref
stores a prefix of the encoded pattern (smaller than
one byte) for all possible alignments a ∈ {0, 1,2,3}.
The variable suf stores a suffix of the encoded pattern
(smaller than one byte) for all possible alignments
a ∈ {0, 1,2, 3}. The examples of the stored prefixes
and suffixes can be seen in Figure 2 as the symbols
preceding/following the red rectangles. Similarly, the
variables prefMask and sufMask store the masks (used
for bitwise-and operation with the corresponding byte
in the encoded text) needed to compare a prefix or the
suffix, respectively of the encoded pattern. The while
cycle (line 23) iterates over only three possible align-
ments. The pref value is stored for the alignments
a ∈ {1, 2,3} (starting from the value 3). The prefix for
the alignment a = 0 is an empty string ε and therefore
it is not stored. The pointer to the array of the suffix
values suf is shifted by the value la and it starts from
the position (la + i) mod 4. In every step of the cy-
cle, the algorithm: (i) stores the corresponding prefix
value to pref array and the corresponding prefix mask
to prefMask array; (ii) stores the corresponding suffix
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
70

Algorithm 1: BADPM preprocessing and searching phase.
1: function BUILDDICTIONARY(P , m)
2: D ←
/
0; B ←
/
0;
3: i ← 0;
4: while i ≤ 3 do
5: b ← b(m − i)/4c×4;
6: E ← ENCODE(P ,i + 1, b);
7: B
i
← E;
8: j ← 1;
9: while j < b(m − i)/4c do
10: for each variant E
j,2
do
11: if D
E
j,2
=
/
0 then
12: D
E
j,2
← create a new list storing offsets and alignments;
13: add a couple of offset and alignment (j −1,i) to the list D
E
j,2
;
14: j ← j + 1;
15: i ← i + 1;
16: function BUILDMASK(P , m)
17: pref ←
/
0; prefMask ←
/
0; prefM ← h0011 1111i;
18: suf ←
/
0; sufMask ←
/
0; sufM ← h1111 1100i;
19: prefB ← ENCODE(P ,1, 4) 2;
20: sufB ← ENCODE(P ,m −4,4) 2;
21: la ← m mod 4;
22: i ← 1;
23: while i ≤ 3 do
24: prefMask
4−i
← prefM;
25: sufMask
(la+i) mod 4
← sufM;
26: pref
4−i
← prefB;
27: suf
(la+i) mod 4
← sufB;
28: prefM ← prefM 2; prefB ← prefB 2;
29: sufM ← sufM 2; sufB ← sufB 2;
30: i ← i + 1;
31: function SEARCH(T , n, P , m)
32: BUILDDICTIONARY(P ,m);
33: BUILDMASK(P , m);
34: shift ← bm/4c −2;
35: i ← shift;
36: while i < n do
37: SYNCVARIANTPOINTERS(i);
38: for each variant T
i,2
do
39: if D
T
i,2
6=
/
0 then
40: for each couple of offset and alignment (o,a) ∈ D
T
i
do
41: SYNCVARIANTPOINTERS(i − o);
42: r ← compare sequentially all bytes starting from T
i−o
with B
a
considering all byte variants;
43: if r = 0 & a 6= 0 then
44: SYNCVARIANTPOINTERS(i −o − 1);
45: r ← compare (T
i−o−1
& prefMask
a
) with pref
a
considering all variants T
i−o−1
;
46: if r = 0 & a 6= la then
47: SYNCVARIANTPOINTERS(i −o + shift);
48: r ← compare (T
i−o+shift
& sufMask
a
) with suf
a
considering all variants T
i−o+shift
;
49: if r = 0 then
50: report an occurrence at position 4 × (i − o)− a + 1;
51: i ← i + shift;
value to suf array and the corresponding suffix mask
to sufMask array; (iii) shifts auxiliary variables prefM
and prefB two bits right; (iv) shifts auxiliary variables
sufM and sufB two bits left.
The function SEARCH represents the main func-
tion of BADPM. After the preprocessing of the searched
pattern (lines 32 and 33) the algorithm states a safe
shift as the number of whole bytes of the encoded pat-
On-line Searching in IUPAC Nucleotide Sequences
71
00 01 00 11 00 00 11 11 00 00 11 01
A C
V
T
A
A T
...
0
4 879
5 903
6 927
dictionary
A → 00
C → 01
G → 10
T → 11
Source sequence
T
A
R T
...
...
...
B
i
B
i+1
B
i+2
Encoded sequence
B
00 01 01 11
00 01 10 11
...
...
00 00 11 10
00 00 11 11
...
...
00 10 11 01
00 10 11 10
00 10 11 11
variants
variants
i
i + 2
3
6
variantPos
variantNum
...
...
...
...
j
j + 1
baseSeq
Preprocessed
pattern
Figure 3: BADPM searching phase. Checking different factors implied from different byte variants.
tern minus two (line 34). The while cycle (line 36)
traverses the encoded text T of length n. It always
reads a byte couple value T
i,2
and all its variants and
the corresponding entries in the dictionary D
T
i,2
are
checked (line 39). The pointers for the array contain-
ing byte variants need to be synchronized for a given
position i − o (line 41). If the dictionary entry D
T
i,2
is
empty the algorithm shifts (line 51) and it continues at
the next position. Otherwise, the algorithm has to tra-
verse over all couples (o, a) stored in the correspond-
ing list and perform three-level comparison for every
couple. The first level is a comparison of the bytes
in the encoded text (starting at the position given by
the offset o) with the bytes of the encoded pattern B
a
according to the shift/alignment a (see line 42). The
second level (see line 45) is a comparison of the prefix
and it is applied only if the first level was successful.
The variant pointers need to be synchronized again
(line 44). The third level (see line 48) is a compari-
son of the suffix and it is applied only if the second
level was successful. If all levels of the comparison
are successful the algorithm reports a new occurrence
at the corresponding position 4 ×(i− o)−a +1 in the
raw text (line 50).
Example 3.1. We state an example to present the way
BADPM processes the degenerate symbols. Suppose
the situation depicted in Figure 3. Current position in
the encoded text is i. The algorithm checks the couple
of bytes B
i
and B
i+1
. The variant pointers are syn-
chronized (line 37 in Algorithm 1) and the algorithm
checks if B
i
or/and B
i+1
contain a degenerate symbol.
The check is evaluated according to the next degener-
ate symbol position in variatPos array. The element
variantPos
j
points to the position i which implies that
B
i
byte has more than one variant. The number of
variants is variantNum
j
= 3. All byte variants are
traversed (line 38 in Algorithm 1 and the red rectan-
gles in Figure 3) and the corresponding entries in the
dictionary are evaluated (lines 39 - 50). The eval-
uation consists of byte-by-byte comparing T
i−o
with
the encoded pattern (with corresponding alignment)
B
a
(line 42). The prefix and suffix comparison must
be performed as well (line 45 and line 48). All byte
variants are considered in the aforementioned com-
parisons.
4 PARALLEL NAIVE SEARCH
We prepared two simple baseline algorithms for
our experiments: Boyer-Moore-Horspool (BMH) algo-
rithm and Parallel Naive Search (PNS). BMH represents
a version of well-known algorithm (Horspool, 1980)
adapted for degenerate strings. However, PNS (thanks
to its engaging results proved in the experiments) de-
serves a little bit deeper description. The idea of PNS
is based on intrinsic parallelism of computer words
and on a bit representation of the alphabet symbols.
All symbols of IUPAC alphabet are represented using
code words with the size four bits (left or right nibble
of a byte). The bits of a code word represent four solid
symbols of the alphabet Σ
solid
= {A,C, G,T } starting
from the least significant bit to the most significant
bit. The bits of the code word are set when the rep-
resented symbol includes the given solid symbol (see
the bit coding in Table 1). Both the processed text T
and the searched pattern P need to be encoded in this
fashion.
PNS performs standard forward pattern matching
process. However,
w
4
symbols are processed at once
in parallel fashion where w is a size of the computer
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
72

Algorithm 2: PNS searching phase.
1: function COMPAREWORDS(a, b)
2: sub ← h0001 0000i
w/8
;
3: mask ← h0000 1111i
w/8
;
4: c ← a & b;
5: r
1
← sub −(c & mask);
6: r
2
← sub −((c 4) & mask);
7: r ← ((r
1
& sub) 1) k (r
2
& sub)
8: function SEARCH(T , n, P , m)
9: pMask ← BUILDPATTERNMASK(P ,m);
10: rMask ← h1100 1111i
w/8
;
11: i ← 1;
12: while i ≤ n −m do
13: j ← 1;
14: r ← h0000 0000i
w/8
;
15: while j ≤ m do
16: r ← r k COMPAREWORDS(T
i+ j,
w
8
,pMask
j
);
17: if (r k rMask) = h11111111i
w/8
then
18: go to shift;
19: j ← j + 1
20: j ← 1;
21: while j ≤ m do
22: r ← r k COMPAREWORDS(T
i+ j,
w
8
4, pMask
j
);
23: if (r k rMask) = h11111111i
w/8
then
24: go to shift;
25: j ← j + 1
26: report all positions/nibbles corresponding to unset bits in r k rMask;
27: shift:
28: i ← i +
w
8
;
word. SEARCH function described in Algorithm 2
represents a basic loop of the algorithm. The param-
eters of the SEARCH function are: an encoded text T ,
a length of the encoded text n, an encoded pattern P ,
a length of the encoded pattern m (the length of the
raw/unencoded pattern is consequently 2m). In the
first step (line 9), the searched pattern P is prepro-
cessed into the form of array pMask. The situation
is depicted in the right part of Figure 4. This array
consists of 2m computer words of length w. Every
element of the array contains a mask composed of
w
4
code words of the corresponding symbol of pattern
P . The first m elements of the array represent the odd
symbols and the last m elements represent the even
symbols of the searched pattern. The variable rMask
represents a register necessary to store the intermedi-
ate results. The register is a computer word composed
of
w
8
bytes. The third and fourth most significant bits
of j-th byte rMask
j
represent the activity of the left
and right nibble of (i + j)-th byte in text T
i+ j
. If the
bit is zero then the corresponding position in the text
is still a candidate for a match. The main loop of
SEARCH function traverses over the encoded text T
(line 12). In every step, a parallel comparison of cor-
responding factor of the text T
i+ j,
w
8
is performed. The
odd symbols of the pattern are compared in the first
block (lines 15–19). The even symbols of the pattern
are compared in the second block (lines 21–25). This
is a technical optimization implying only one shifting
of the compared text factor T
i+ j,
w
8
. Whenever no ac-
tive position remains (no zero bit in rMask, lines 17
and 23), the comparing sequence stops and the text is
shifted (line 28).
The parallel comparison of the text factor T
i+ j,
w
8
with pMask
j
is done in function COMPAREWORDS.
This function is performed in constant time and its
usage is depicted in Figure 4. The bitwise AND
operation between the text factor T
i+ j,
w
8
and the
corresponding pMask
j
is the first step (line 4).
Next, the non-empty intersection in terms of single
nibbles need to discovered. The right nibbles are
checked at first. The intermediate results of bitwise
AND operation is masked and then subtracted from
sub register where only the fourth most significant
bit is set (line 5). If the bit is set even after the
subtraction it means that there is an empty inter-
section (corresponding symbols do not match) and
the corresponding position in the text is excluded
On-line Searching in IUPAC Nucleotide Sequences
73

A C
V
T
...
Source sequence
A
R T
...
Encoded sequence
B
0001 0010
...
0111 1000
...
...
0001 0101
1000 1110
...
2i
2i+15
i
i+7
Preprocessed pattern
0001 0001 ... 0001 0001
Pattern
A C A T ... A A T C
0010 0010 ... 0010 0010
0001 0001 ... 0001 0001
1000 1000 ... 1000 1000
...
0001 0001 ... 0001 0001
1000 1000 ... 1000 1000
0001 0001 ... 0001 0001
1
|w|
1
m
0001 0000
0001 0000
0001 0001
0000 0000
...
&
0000 1111 ... 0000 1111
mask
0001 0000 ... 0001 0000
sub
&
−
0000 0000
0000 0000
0000 0001
0000 0000
...
0001 0000
0001 0000
0000 1111
0001 0000
...
&
0001 0000
0001 0000
0000 0000
0001 0000
...
r
1
0010 0010 ... 0010 0010
...
m + 1
2m
Figure 4: PNS searching phase. The first step of comparing the source sequence at position 2i with the preprocessed pattern.
from the potential candidates for a match. The same
operation is performed for the left nibbles (line 6)
and the intermediate results are merge into one
register r (line 7). The searching phase of PNS clearly
requires O(
n
w
m) time at most where w is a size of
the computer word. Very simple preprocessing of the
pattern is required and it takes O(m) time.
Example 4.1. We demonstrate the function COM-
PAREWORDS in the following example (see Figure 4).
Suppose text factor T
2i,16
with a prefix “ACVT” and
a suffix “ARTB”. Furthermore, suppose a pattern P
with a prefix “ACAT” and a suffix “AATC”. After the
bitwise AND operation between the encoded text fac-
tor and the corresponding mask pMask
1
only four of
the depicted nibbles have non-empty intersection: the
first symbol ‘A’, the third symbol ‘V’, the first sym-
bol of the suffix ‘A’ and the second symbol of the suf-
fix ‘R’. The active bits are emphasize with red color.
Next, the comparison of the right nibbles is performed
and the corresponding bitwise AND operation with
the register mask keeps active only the second nibble
of the suffix (symbol ‘R’). Subtraction is performed
and the corresponding bit (fourth most significant bit
in the first byte of the suffix) stays unset which implies
that the corresponding position 2i + 13 in the text is
still a candidate for a match. The processing of the
left nibbles is skipped to simplify the example.
5 EXPERIMENTS
We present experimental results that give a detailed
comparison of our algorithm BADPM with the afore-
mentioned baseline algorithms. Particularly, we per-
formed the comparison in terms of locate time with
Boyer-Moore-Horspool (BMH) algorithm (Horspool,
1980), with Parallel Naive Search (PNS) algorithm
(see Section 4) and with Backward Nondeterminis-
tic DAWG Matching (BNDM) algorithm (Navarro and
Raffinot, 1998) optimized for IUPAC alphabet. We
planned also a comparison with the algorithm pro-
posed by Iliopoulos in (Iliopoulos et al., 2008). How-
ever, the authors did not provide an implementation
of their algorithm. The essence of BADPM (its byte ori-
entation and its principle of tabulating all factors) pre-
determines this algorithm to search for patterns with
length m ≥ 12 bases. BMH and BNDM shall also profit
from the longer searched patterns, while PNS is prac-
tically independent of the length of the searched pat-
tern m. Theoretically, PNS locate time could worsen
with the growing pattern length thanks to its time
complexity O(
n
w
m), however, the experiments did not
prove this trend.
All the tested algorithms were implemented in C
programming language
3
. We carried out our tests on
Intel
R
Core
TM
i7-4702MQ 2.20 GHz, 8 GB RAM.
We used compiler gcc version 5.4.0 with compiler
3
BADPM implementation is available at http://www.
stringology.org/badpm/badpm.zip
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
74

0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
0 200 400 600 800 1000
Locate time [sec]
Pattern length
BADPM SO64 BMH BNDM
Figure 5: Human chromosome 7: Locate time depending
on the length of the searched pattern m.
optimization -O3. The tested patterns were chosen
randomly from the input text and their length m was
ranging from 16 to 1024. All experiments were run in
loop 1 000 times and we report the mean of the run-
ning time in seconds. All reported times represent
measured user time + sys time and they always in-
clude any necessary pattern preprocessing. For eval-
uating the algorithms, we used vcf files of differ-
ent chromosomes downloaded from 1000 Genomes
Projects (Consortium, 2011)
4
. The vcf files were
transformed into the consensus text files over IUPAC
alphabet using bcftools consensus utility by Sanger
institute with parameter -I denoting the output given
in IUPAC alphabet.
The first experiment describes dependency of lo-
cate time on the length of the searched pattern for
single algorithms. BADPM proves strongly improving
locate time when the length of the pattern P grows
up to the value 256 bases. This is expected result
since the shift of BADPM basic loop directly depends
on the length of the pattern. BMH also derives its shift-
ing from the pattern length. However, its improve-
ment is not so strong thanks to small alphabet size
and the degenerate string domain which limits shift-
ing potential of BMH. BNDM should also profit from the
increasing length of the searched pattern, however, it
is limited by the size of the computer word. Since the
length of the pattern exceeds the size of the computer
word BNDM shows no further improvement. BADPM
proved its superiority in locate time for middle-sized
and long patterns. BADPM is almost three-times faster
than BMH for m = 16. PNS and BNDM achieve only a
slightly worse locate time than BADPM for m = 16.
However, BADPM strongly dominates for longer pat-
terns. It substantially improves its locate time until
4
ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130
502/
the pattern length m = 256 when the practical lower
bound is achieved.
We compared the locate time of single algorithms
on real genomic data obtained from 1000 Genomes
Projects (Consortium, 2011) in our second experi-
ment (see Figure 6). We chose a middle size of the
searched pattern (m = 16) which implies no signifi-
cant shifting benefit for BADPM which dominates es-
pecially for longer patterns. Still, BADPM achieved
the best locate time for all tested chromosomes. PNS
thanks to its parallelism performs a relatively large
shifts corresponding to the computer word size w and
so it achieves also very good locate time over all
tested files. BNDM proved also very good results since
m = 16 is clearly lower than the size of the computer
word. The second vertical axis in Figure 6 reports the
size of the chromosome files. The experiment proves
that the locate time of all tested algorithms directly
corresponds to the size of the tested files.
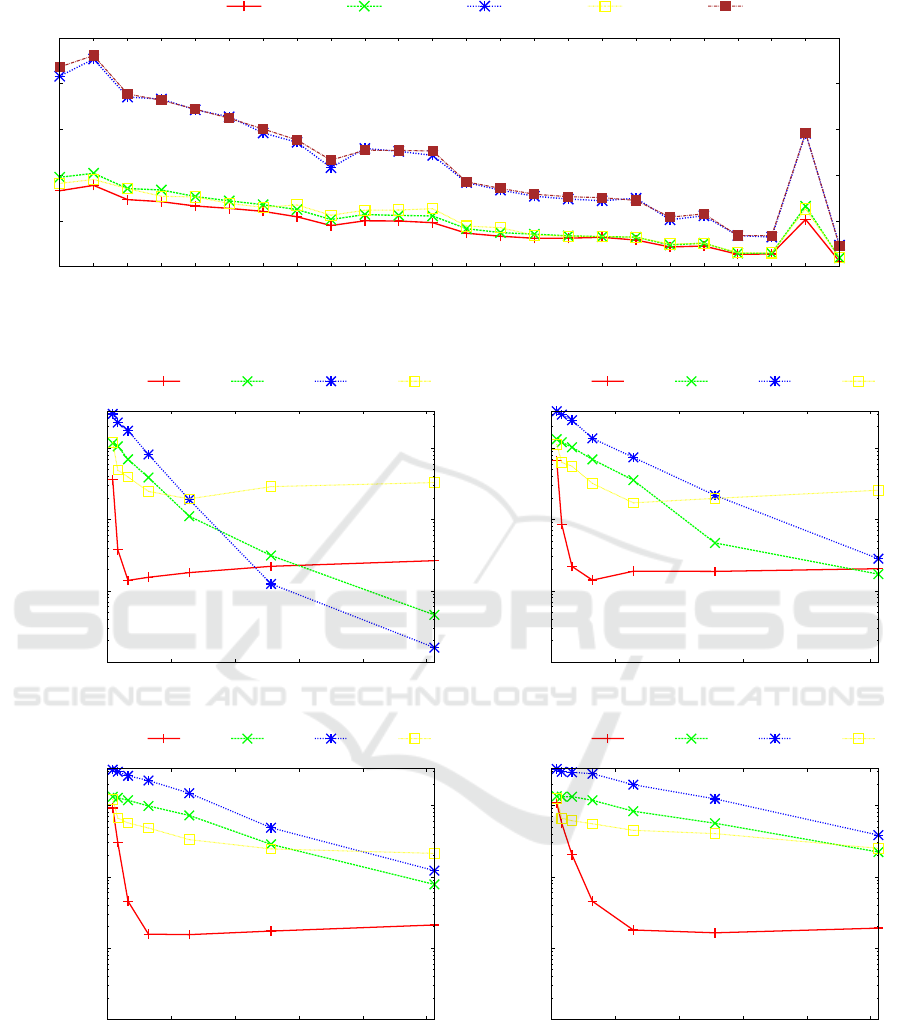
We combined all tested algorithms with a simple
q-gram block inverted index defined in Section 2 to
achieve locate time that is able to compete with the
times achieved by other index data structures (e.g.
self-indexes). We tested all algorithms for different
block sizes (varying from 12800 to 102 400 bases) of
the q-gram inverted index (see Figure 7). We can ob-
serve that for smaller block sizes (12800 and 25 600
bases, see Figure 7(a) and Figure 7(b)) and for longer
patterns (m ≥ 512 bases) the algorithms with simpler
preprocessing phase (PNS and BMH) dominate. The
reason is that the inverted index performs very effi-
cient filtration of the blocks for longer patterns and
smaller blocks. Thus, the following search algorithm
itself processes only a small portion of the file and
so the time needed to preprocess the searched pat-
tern dominates over the search time of the algorithm.
The vertical axis in Figure 7 are given in logarithmic
scale to achieve a better overview of the achieved re-
sults. The minimal locate times achieved generally
for the longer patterns are less than one millisecond.
This locate time is two orders of magnitude worse
than the time needed for standard (not degenerate)
pattern matching problem (see Experiments section
in (Proch
´
azka and Holub, 2017)). However, the time
around one millisecond is still competitive for many
applications.
6 CONCLUSION AND FUTURE
WORK
We proposed the algorithm BADPM optimized for
searching in the degenerate text over an alphabet of
a small size. Practically, BADPM is designed to search
On-line Searching in IUPAC Nucleotide Sequences
75
0
0.1
0.2
0.3
0.4
0.5
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X Y
0
50
100
150
200
250
Locate time [sec]
File size [MiB]
Chromosome
BADPM SO64 BMH BNDM File size
Figure 6: Locate time for different human chromosomes for m = 16. The second vertical axis represents the chromosome file
size.
0.0001
0.001
0.01
0.1
0 200 400 600 800 1000
Locate time [sec]
Pattern length
BADPM SO64 BMH BNDM
(a) Block size = 12 800
0.0001
0.001
0.01
0.1
0 200 400 600 800 1000
Locate time [sec]
Pattern length
BADPM SO64 BMH BNDM
(b) Block size = 25 600
0.0001
0.001
0.01
0.1
0 200 400 600 800 1000
Locate time [sec]
Pattern length
BADPM SO64 BMH BNDM
(c) Block size = 51 200
0.0001
0.001
0.01
0.1
0 200 400 600 800 1000
Locate time [sec]
Pattern length
BADPM SO64 BMH BNDM
(d) Block size = 102 400
Figure 7: Human chromosome 7: Locate time depending on the length of the searched pattern m. The vertical axis is in
logarithmic scale.
very efficiently in pangenomic data of a population
of the same species, specifically vcf files. BADPM is
based on the idea of BAPM (Proch
´
azka and Holub,
2017) and it inherits its basic properties: (i) process-
ing at byte level of the input text; (ii) tabulating all
possible factors of the pattern and searching for them
in the filtration step. These two properties ensure
very competitive locate time in the order of millisec-
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
76

onds and sublinear average time complexity. This was
proven on real pangenomic data from 1000 Genomes
Projects (Consortium, 2011).
We plan to design search algorithms for other
forms of pangenomic data in our future work. This
includes the algorithms optimized for so-called Elas-
tic Degenerate Strings which is another form of rep-
resentation genomic data for a population of the same
species.
ACKNOWLEDGEMENTS
The research was partially supported by OP
VVV project Research Center for Informatics no.
CZ.02.1.01/0.0/0.0/16 019/0000765.
REFERENCES
Baeza-yates, R. A. (1992). Text retrieval: Theory and prac-
tice. In In 12th IFIP World Computer Congress, vol-
ume I, pages 465–476. Elsevier Science.
Bernardini, G., Pisanti, N., Pissis, S. P., and Rosone, G.
(2017). Pattern matching on elastic-degenerate text
with errors. In Fici, G., Sciortino, M., and Ven-
turini, R., editors, String Processing and Information
Retrieval, pages 74–90, Cham. Springer International
Publishing.
Boyer, R. S. and Moore, J. S. (1977). A fast string searching
algorithm. Commun. ACM, 20(10):762–772.
Cisłak, A., Grabowski, S., and Holub, J. (2018). Sopang:
online text searching over a pan-genome. Bioinfor-
matics, page bty506.
Consortium, T. . G. P. (2011). A map of human genome
variation from population-scale sequencing. Nature,
473:544 EP –. Corrigendum.
Consortium, T. U. (2015). The uk10k project identifies rare
variants in health and disease. Nature, 526:82 EP –.
Crochemore, M., Iliopoulos, C. S., Kundu, R., Mohamed,
M., and Vayani, F. (2015). Linear algorithm for
conservative degenerate pattern matching. CoRR,
abs/1506.04559.
Crochemore, M. and Rytter, W. (1994). Text Algorithms.
Oxford University Press, Inc., New York, NY, USA.
D
¨
om
¨
olki, B. (1964). An algorithm for syntactical analy-
sis. Computational Linguistics, 3:29–46. Hungarian
Academy of Science, Budapest.
Grossi, R., Iliopoulos, C. S., Liu, C., Pisanti, N., Pissis,
S. P., Retha, A., Rosone, G., Vayani, F., and Versari, L.
(2017). On-line pattern matching on similar texts. In
28th Symposium on Combinatorial Pattern Matching,
CPM 2017, July 4-6, 2017, Warsaw, Poland, pages
9:1–9:14.
Holub, J., Smyth, W., and Wang, S. (2008). Fast pattern-
matching on indeterminate strings. Journal of Dis-
crete Algorithms, 6(1):37 – 50. Selected papers from
AWOCA 2005.
Horspool, R. N. (1980). Practical fast searching in strings.
Software: Practice and Experience, 10(6):501–506.
Iliopoulos, C. S., Kundu, R., and Pissis, S. P. (2017). Ef-
ficient pattern matching in elastic-degenerate texts.
In Drewes, F., Mart
´
ın-Vide, C., and Truthe, B., ed-
itors, Language and Automata Theory and Applica-
tions, pages 131–142, Cham. Springer International
Publishing.
Iliopoulos, C. S., Mouchard, L., and Rahman, M. S. (2008).
A new approach to pattern matching in degenerate
DNA/RNA sequences and distributed pattern match-
ing. Mathematics in Computer Science, 1(4):557–569.
Knuth, D. E., Morris, J. H., and Pratt, V. R. (1977). Fast
Pattern Matching in Strings. SIAM Journal on Com-
puting, 6(2):323–350.
Manber, U. (1997). A text compression scheme that allows
fast searching directly in the compressed file. ACM
Trans. Inf. Syst., 15(2):124–136.
Marschall, T. (2018). Computational pan-genomics: status,
promises and challenges. Briefings in Bioinformatics,
19(1):118–135.
Navarro, G. and Raffinot, M. (1998). A bit-parallel ap-
proach to suffix automata: Fast extended string match-
ing. In Proceedings of the 9th Annual Symposium
on Combinatorial Pattern Matching, CPM ’98, pages
14–33, London, UK, UK. Springer-Verlag.
Navarro, G. and Raffinot, M. (2002). Frontmatter, pages
i–iv. Cambridge University Press.
Proch
´
azka, P. and Holub, J. (2017). Byte-aligned pattern
matching in encoded genomic sequences. In 17th
Int. Workshop on Algorithms in Bioinformatics, WABI
2017, August 21-23, 2017, Boston, MA, USA, pages
20:1–20:13.
Puglisi, S. J., Smyth, W. F., and Turpin, A. (2006). Inverted
Files Versus Suffix Arrays for Locating Patterns in Pri-
mary Memory, pages 122–133. Springer Berlin Hei-
delberg, Berlin, Heidelberg.
Sunday, D. M. (1990). A very fast substring search algo-
rithm. Commun. ACM, 33(8):132–142.
Wu, S. and Manber, U. (1992). Agrep - a fast approximate
pattern-matching tool. In In Proc. of USENIX Techni-
cal Conference, pages 153–162.
On-line Searching in IUPAC Nucleotide Sequences
77
