
A Proposal for a Language Combining Biochemical Rules and
Topological Structure for Systems Biology
Anasthasie Joelle Compaore
1 a
and Pascale Le Gall
2 b
1
Ecole Supérieure d’Informatique, Université Nazi Boni, Bobo-Dioulasso, Burkina Faso
2
Laboratoire MISC, CentraleSupélec, Université Paris Saclay, Gif-Sur-Yvette, France
Keywords:
Rule-Based Modelling, Biochemical Rules, Compartments, Topology-Based Modelling.
Abstract:
For about twenty years, rule-based modelling has been widely used for Systems Biology issues. Most existing
languages focus on biochemical reactions primarily, and to a lest extent, on the cell structure in compartments.
BIOCHAM and Pathway Logic Assistant (PLA) are representative examples of such rule-based languages.
They are equipped with tools providing great analysis capabilities. We propose to provide such biochemical
languages with annotations relating to the compartments in which the biochemical reactions take place. We
will make sure that biochemical rules always indicate the nature of the compartments involved and the neigh-
bourhood relations between them. At the end, it suffices to specialize the generic rules according to a particular
topological structure in order to obtain sets of localized rules. Thus, resulting models can be analysed by using
either BIOCHAM or PLA.
1 INTRODUCTION
Systems biology concerns the study of molecular in-
teraction networks at the cellular level by using differ-
ent disciplinary fields such as biochemistry, cell biol-
ogy, computer science, mathematics or systems en-
gineering. Cellular biological mechanisms are con-
sidered as natural complex systems (Ma’ayan, 2017)
mainly characterized by unpredictability, context de-
pendency, emergence and stochasticity (Chen and
Crilly, 2016). A lot of aspects participate to this com-
plexity: the dynamic rearrangements of the compart-
ments, related to the movements of membranes; the
kinetic parameters characterizing each reaction; the
combinatorial complexity due to the possible great
numbers of the states of the molecules knowing that
for a given molecule M, different states of M can
induce different functionalities and thus different re-
actions involving M. Because of this intrinsic com-
plexity, elucidating or predicting the functioning of
biological systems generally proceeds by modelling.
This modelling aims at interconnecting the different
underlying cellular processes - at the cellular level
- and integrating different hierarchies of cells - at a
cells population level. In this effort, a lot of modelling
a
https://orcid.org/0000-0003-3045-9579
b
https://orcid.org/0000-0002-8955-6835
approaches, based on a diversity of formalisms (Bar-
tocci and Lio, 2016)) have been defined and used in
the study of some phenomenons such as signal trans-
duction (Talcott, 2016; Riesco et al., 2017), gene reg-
ulation (Faeder et al., 2009), metabolism and protein-
protein interactions (Fages et al., 2004).
The way the coupling between biochemical reac-
tions and compartmentalization is made differs from
one approach to another: while the spatio-temporal
dynamics-oriented approaches explain the interested
phenomenon only by the dynamic of the system’s ge-
ometry (Regev et al., 2004; Cardelli, 2004; Gian-
nakis and Andronikos, 2017), some others such as
BIOCHAM (Fages et al., 2004), pathway Logic (Eker
et al., 2002), the first version of BioNetGen (Faeder
et al., 2005) focus on the biochemical reactions in an
implicit static compartmentalization. The majority of
the first models are equation-based and permit quan-
titative analysis. However, these modellings can be
limited by the number of equations to be defined (due
to the combinatorial complexity) and the difficulties
in the estimation of kinetics. Therefore, other for-
malisms, inspired by computer science such as pro-
cess calculi, cellular automata, agents and rule-based
languages have been used, and the so designed mod-
elling tools permit quantitative and/or qualitative sim-
ulations and analysis (Machado et al., 2011; Pedersen
et al., 2015).
310
Compaore, A. and Gall, P.
A Proposal for a Language Combining Biochemical Rules and Topological Structure for Systems Biology.
DOI: 10.5220/0007689303100317
In Proceedings of the 12th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2019), pages 310-317
ISBN: 978-989-758-353-7
Copyright
c
2019 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
We chose to focus on rule-based modelling of bio-
chemical reactions in a static compartmentalization
for the following main reason: a rule-based modelling
formalism is generally considered as relatively sim-
ple and intuitive: a reaction results in changes in the
contents - in terms of present molecules and/or their
quantities or concentrations - of some compartments
of the considered system. Rules, representing reac-
tions, are then of the form Le f t Hand → Right Hand
to materialize this transition of states. Le ft Hand
and Right Hand respectively represent the needed
molecules for the reaction to occur and the molecules
resulting from this occurrence. This form of rules
is familiar to biologists because inspired of classi-
cal chemical equations. To simplify modelling a lit-
tle more, molecules are often abstracted by identi-
fiers and if necessary, mechanisms are added to spec-
ify sites. For a very classical example, the reac-
tion of water dislocation can be expressed by the rule
2A −→ B +C where A, B et C respectively stand for
molecules H
2
O, H
3
O
+
and HO
−
. In the context of bi-
ology, the localization of the involved molecules is a
key information to be added to this rule. In (Hlavacek
et al., 2006), one can find other advantages of rule-
based languages for the modelling of biological cells.
The remainder of the paper is organized as follows: in
Section 2, we present in more details our motivations
in the context of rule-based languages for systems bi-
ology. In Section 3, we outline our approach empha-
sizing the role of compartmentalization and the inter-
est of systematically locating biomolecules in com-
partments in a generic way. In Section 4, we illustrate
our approach on some examples issued from the liter-
ature. Section 5 gives some concluding remarks.
2 MOTIVATIONS
2.1 Context
Rule-based modelling is widely used in Systems Bi-
ology as evidenced by the important number of tools
such as Kappa (Danos and Laneve, 2004; Boutil-
lier et al., 2018), Virtual Cell (VCell) (Blinov et al.,
2017) and BioNetGen (Faeder et al., 2009) issued
from studies dealing with the explicitation of biolog-
ical phenomena by the mean of this formalism. The
rule-based languages underlying these three tools are
designed for the definition of models that especially
track biomolecular sites dynamics. Generally, the ex-
pressible reactions are reversible and consist in the
synthesis of a molecule, the binding of two molecules
and the modification of the form of a molecule. Those
of the languages that take into account compartments
can express the transport of a molecule from one com-
partment to another and species spanning multiple
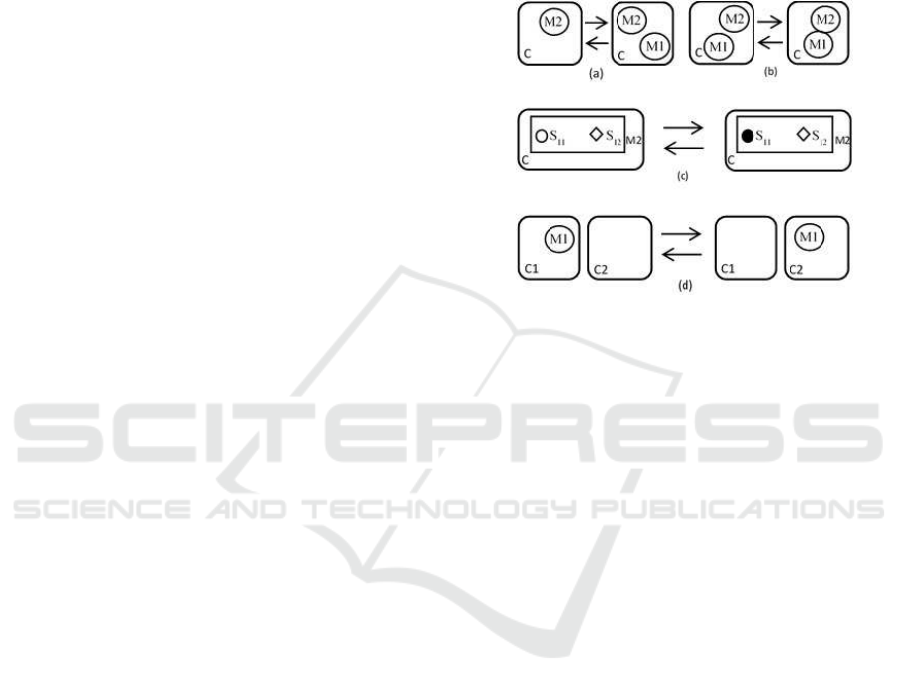
compartments. Figure 1 gives a schematic representa-
tion of some localized reactions. More or less similar
schematics can be found in the literature of most of
the aforementioned languages and their understand-
ing is intuitive.
Figure 1: Representation of some rules. (a): Synthesis (→)
and Degradation (←) of M1 catalysed by M2 in C. (b):
Binding (→) and Unbinding (←) of M1 and M2 in C. (c):
Phosphorylation (→) and Dephosphorylation (←) of M2 on
site S
11
in C. (d): Transport of M1 from C1 to C2 (→) and
from C2 to C1 (←).
Each of the tool-sets provide capabilities to simulate,
analyse and visualize networks generated from the
models and the graphs issued from simulation. Cur-
rently, more and more initiatives are working to en-
able modellers to easily take advantage of the func-
tionality of different tools. That results either in trans-
lators like the Kappa-BioNetGen one introduced by
(Suderman and Hlavacek, 2017) that enables transla-
tion in the two senses or in provided capabilities of
inferring from a given model, another model based
on a different formalism.
Most of the rule-based approaches first focused on
biochemical reactions before proposing extensions to
highlight the topology a little more. BioNetGen Lan-
guage (BNGL) has therefore been extended, result-
ing in compartmental BNGL (cBNGL) (Harris et al.,
2009) that makes it possible to explicit topology as an
inclusion graph. Similarly, the modeling of compart-
ments and space within the cell is possible in Virtual
Cell (VCell) since the extension presented in (Blinov
et al., 2017).
The approach we propose in this paper depends on
BIOCHAM (Fages and Soliman, 2008) and Path-
way Logic (PL) (Eker et al., 2002; Talcott, 2016).
BIOCHAM is an environment to modelling biochem-
ical interactions which provides a rule-based lan-
guage for the definition of the models and a tempo-
A Proposal for a Language Combining Biochemical Rules and Topological Structure for Systems Biology
311

ral logic based language to express properties that are
supposed to be checked by the system. It offers differ-
ent semantics, the boolean one being based on rewrit-
ing logic, and the carrying out of both qualitative and
quantitative analysis. It also enables the checking
(and the learning (Calzone et al., 2005)) of the prop-
erties of the models by the using of NuSMV model-
checker. BIOCHAM has been extended to provide the
capability of infering rule-based models from ODE-
based one (Fages et al., 2015). This is useful when
kinetics parameters value are not totally defined. PL
is another rule-based approach to modelling cellular
processes, that uses the rewriting language MAUDE
(Clavel et al., 2000) to write the algebraic specifica-
tion underlying its models. It queries its models by
using Pathway Logic Assistant (PLA) (Talcott and
Dill, 2005) that offers a graphical interface and ac-
cesses to some formal tools such as Pathanalyser (Dill
et al., 2005) and the simulation and model-checking
features of MAUDE. These tools materialize localiza-
tions by attaching simple labels to molecules, with a
difference for PL that can write models from which
one can deduce the hierarchical organization of cells
found in Biomodels (Li et al., 2010). A model in these
languages is not explicitly guaranteed to be coherent
with a specific topology. It is this aspect of combin-
ing rules with formal topological structure that we ad-
dress in this paper.
2.2 Motivations
Living organisms are highly compartmentalized bio-
logical systems whose functioning depends on the co-
ordination of the isolated behaviours of each compart-
ment. Then, topology, i.e the nature of compartments
composing the system and their relative position in
the system, as well as biochemical reactions are de-
terminant for the life of the system. The importance
of topology is directly related to the role of compart-
mentalization. On one hand, compartments of same
nature are entities sharing the same functionalities and
the same behaviour. On the other hand, by group-
ing in each compartment the appropriate molecules
(due to the selective permeability of membranes),
it improves systems efficiency by speeding up the
molecular interactions given that their occurrences
are location-dependant. This role of compartmen-
talization is highlighted by (Harris et al., 2009) that
presents a compartmental model of an eukaryotic cell
in which a signalling process results from interactions
between molecules from four distinct compartments
each containing specific molecules and transport re-
actions concerning neighbouring compartments.
Despite its importance, a relative weakness in the tak-
ing into account of the topology can be noticed in
most formalisms for rule-based modelling of biolog-
ical processes. Our motivation is then to reinforce
in these modellings, the place of the topology by en-
abling its separate representation. A particular regard
is put on the neighbouring relationships to take into
account the location-dependency of reactions, and on
the types of compartments to respect the specific po-
tential behaviour of each compartment. Moreover, the
organels are not abstracted by a generic one (if a sys-
tem has n compartments of type T , all the n compart-
ments are represented), to make possible the simul-
taneous occurrences of concurrent reactions. The re-
sulted models are guaranteed to be coherent with the
static topological structure of the system under con-
sideration: all the rules are contextualized with regard
to the compartments. Then, our models can be used
through existing tools. We have chosen BIOCHAM
and PL as privileged target tools because they have a
lot of similarities.
3 THE MODELLING APPROACH
3.1 General Overview
Classically, in rule-based modelling of biological pro-
cesses, topology and biochemical reactions are taken
into account in an integrated manner. Unlike these ap-
proaches, our rule-based modelling approach focuses
on an explicit specification of the topological struc-
ture. We propose to separate the two following com-
plementary aspects: a static topology of the system
and biochemical reactions. Our approach can then be
considered as a topology-driven rule-based modelling
approach as shown in Figure 2. We propose to take
as input data firstly a graph, that we call as exchange
graph and that abstracts the topology of the system
and secondly, a set of generic rules that describe the
biochemical reactions that can occur in the system
provided that there is some correspondence between
compartments mentioned in the topological structure
and indications of localization w.r.t types of compart-
ments given in the rules. Then, we automatically elab-
orate a concrete model which can be translated in such
a way that it can be handled by the target tool chosen
by the modeller.
3.2 The Exchange Graph
Generally speaking, a compartment of a biological
system is of a certain type and is delimited by a
double-layered membrane. Molecules may be local-
ized more precisely in one of the four locations related
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
312
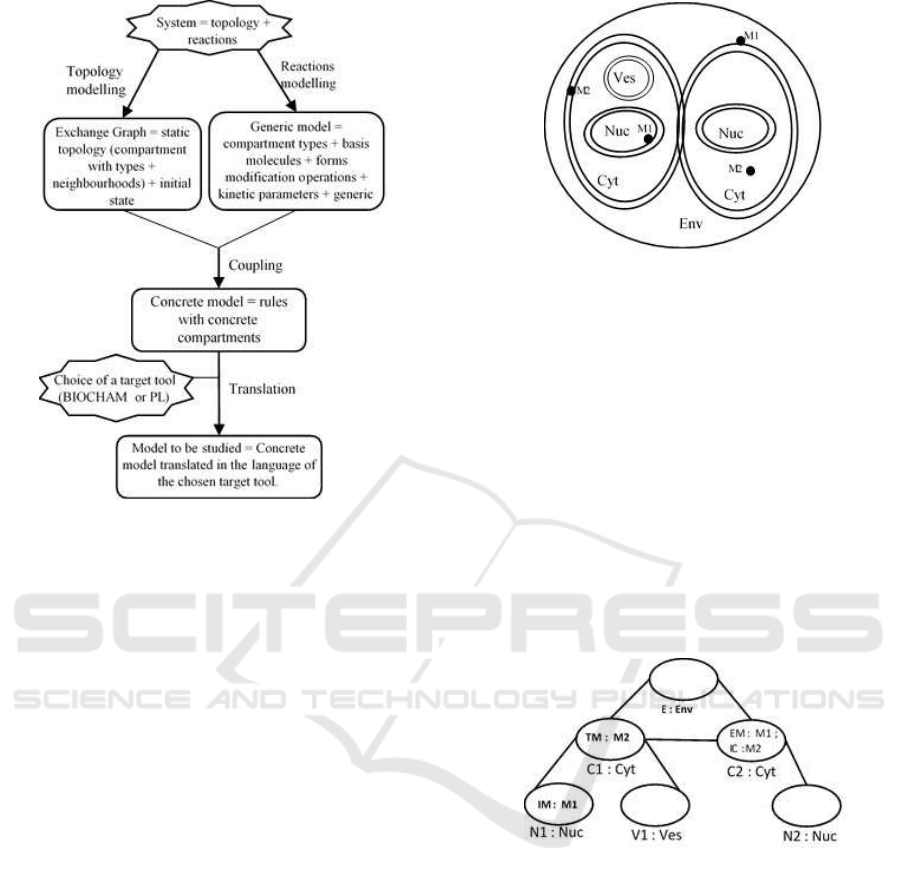
Figure 2: Schematic representation of the approach.
to a given compartment: from the most external to the
most internal, we name these locations EM (molecule
is tethered to the external membrane), T M (molecule
is in the trans-membrane space), IM (molecule is teth-
ered to the internal membrane) and IC (molecule is in-
side the compartment). IC is the default location for
a given compartment. Two kinds of neighbourhoods
for localisations are considered: the neighbourhood
by inclusion in which one compartment is included
in another one and the neighbourhood by adjacency
in which two compartments stuck each other by por-
tions of their membranes. Figure 3 is an example of
such a topology. The particular compartment of type
Env stands for the outer compartment of any system.
Its role is to delimit the system of interest. Gener-
ally speaking, the rules will bring into play molecules
that are in localities connected by neighbourhood re-
lations.
The exchange graph of a biological system S captures
all the neighbourhood relations between the compart-
ments of S and only these ones. It will also store the
contents of these compartments. We note V
S
the set of
the compartments of S, T
S
the set of the types of the
compartments of S and E the unique external com-
partment of type Env. The exchange graph GE
S
as-
sociated to S is an undirected graph in which a ver-
tex represents an element of V
S
∪ E and an edge
represents either the adjacency between two compart-
ments, or the inclusion of a compartment in another
one. Each compartment is associated with its type,
an element of T
S
∪ Env and its content in terms of
Figure 3: A topological structure S
1
. It has two adjacent
compartments of type Cyt: each of them contain a com-
partment of type Nuc while only one of them contains in
addition a compartment of type Ves (for vesicle). The local-
izations of molecules are T M for the first compartment of
type Cyt, EC and IC for the second one and IM for the first
compartment of type Nuc.
molecules. To sum up, we impose the following con-
ditions on the exchange graphs: i) exactly one com-
partment is of type Env and ii) for each element t of
T
S
, there is at least one element of V
S
whose type is
t. For example, when considering the topology S
1
(Figure 3), we have V
S
1
= {C1,C2, N1, N2,V 1} and
T
S
1
= {Cyt, Nuc,Ves} by naming C
1
and C
2
. . . the
compartments of S
1
. Figure 4 gives the schematic rep-
resentation of GE
S
1
.
Figure 4: Exchange Graph GE
S1
for to S
1
(Figure 3).
In practice, there are essentially two ways for obtain-
ing an exchange graph: the first one is to directly
construct it by manually designing it while respect-
ing basic topological and biological conditions. This
method is clearly sufficient when the topology to be
abstracted is simple enough not to be mistaken.
The second way consists in extracting the graph from
geometric models, called the bio-geometric mod-
els and handled by some geometric 3D-modellers.
For our part, we build bio-geometric models us-
ing a specialization of MOKA (Vidil et al., 2002),
a topology-based geometric modeller implementing
3-Generalized-Maps. For modelling an object as
Generalized-Map, the topology of the object is first
modelled by as a set of darts (obtained by decompos-
ing the objects along their topological structure until
A Proposal for a Language Combining Biochemical Rules and Topological Structure for Systems Biology
313

getting elements of smaller dimensions, called darts).
Then embeddings, that is pieces of information such
as forms, colors, dimensions or molecule concentra-
tions, . . . are associated to main elements (such as vol-
umes, faces, vertices) composing the topology in or-
der to obtain a full geometric object.
In our specialization of MOKA, we represent com-
partments as double-cubes (representing respectively
the inner membrane and the outer membrane of the
compartment) linked by a 3-dimensional neighbour-
hood relation. A compartment has then forty-eight
darts (eight for each face). In our setting, the embed-
dings consist in the name, the type and the content of
compartment attached to the 3-cells (volumes) of the
topology. In addition to the creation of the environ-
ment, we offer two other creation operations: i) the
creation of a compartment C1 in a compartment C2 :
the name C2 is associated to the outer membrane of
C1; ii) the creation of a compartment C1 stuck to a
compartment C2 : the bonding surface on C2 must be
defined and 3-dimensional links are established be-
tween corresponding surfaces of C1 and C2. An il-
lustration of the bonding surface is given by Figure 5
where the 3-dimensional links are the horizontal black
lines between two darts (points).
Figure 5: Bonding surface of two volumes.
Figure 6 represents the bio-geometric model of S
1
(Figure 3). Five compartments are visible and we use
colors for distinguishing compartments.
Figure 6: Bio-Geometric model of S
1
(Figure 3).
Using geometric modellers becomes particularly con-
venient for situations involving a significant number
of compartments (e.g. Figure 7)
The extraction of the exchange graph is done by
browsing the darts of the bio-geometric model. For a
compartment C1, the set of its neighbours contains all
Figure 7: Bio-Geometric model of a system that has a com-
partment (the central one) which has no direct neighbour-
hood with its container, because it is adjacent on all its faces
to another compartment.
the compartments which have at least one dart linked
with a dart of C1 by an 3-dimensional link.
3.3 The Generic Model
The generic model is the transcription in our rule-
based language, of the reactions. As shown by Listing
1 that gives an extract of the grammar of the language,
a generic model (GM) is organized in five sections
(line 1) each delimited by BEGIN_section-name and
END_section-name.
The first section CTypes that lists the compartment
types is original to our approach while the others re-
spectively listing the basis molecules (BMols), the
molecules forms transformations (Modifs), the kinetic
parameters identifiers (KPS) and the generic reaction
rules (GRS ) are classically found in the other lan-
guages. The syntax allows association of particular
sites to a basis molecule (lines 8 and 9) and kinetic
parameters are valued (line 16).
Listing 1: Syntax of generic model.
1 GM : : = CTypes BMols Modifs KPS GRS
2
3 CTypes : : = BEGIN_CTypes LCT END_CTypes
4 LCT : : = idCT | idCT ;LCT
5
6 BMols : : = BEGIN_BMols LBM END_BMols
7 LBM : : = BM | BM; LBM
8 BM : : = idBM | idBM ( L S i t e s )
9 L S i t e s : : = i d S i t e | i d S i t e , L S i t e s
10
11 Modifs : : = BEGIN_Modifs LMod END_Modifs
12 LMod : : = idMod | idMod , LMod
13
14 KPS : : = BEGIN_KPS LKP END_KPS
15 LKP : : = KP | KP , LKP
16 KP : : = idKP ( r e e l )
17
18 GRS : : = BEGIN_GRS LGR END_GRS
19 LGR : : = GR | GR LGR
20
21 GR : : = idGR : PREC KParam
22 S t a t e => S t a t e
23 CondMolVar .
24 S t a t e : : = [ ] @CVar | LocSol
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
314

25 LocSol : : = [ MolSet ]@CVar |
26 [ MolSet ]@CVar & LocSol
27 MolSet : : = Molec | Molec + MolSet
28 Molec : : = idBM | ( Molec : Modif ) |
29 Molec−Molec
30 Modif : : = idMod < LS it es > | idMod
31 CVar : : = idCV : idCT TCPart | idCV |
32 idCT TCPart | AnyW
33 TCPart : : = (EM) | (TM) | ( IM) | ( IC ) | _
34 PREC : : = [ PreC : LCond ]
35 LCond : : = {CVar , CVar} | {CVar , CVar}LCond
The generic rules language is widely inspired from
BIOCHAM and PL in the sense that it has been de-
signed to be able to express what is expressible in
the rule-based languages of these environments. List-
ing 1 gives from line 21 the syntax of (the transition
part of) a generic rule which consists in two manda-
tory components (rule identifier (idGR) and transition
part (line 22)), the others being optional. A molecule
(Molec is a basis one, an altered form of a molecule
or a complex). The localizations of molecules are
expressed using @ (line 24 to 26) that attaches a
compartment variable (CVar) to a set of molecules
(solution, MolSet ) or to [] (empty solution). The
compartment-variable gives the type of compartment
in which the molecules are localized. It can be ab-
breviated (line 30, options 2 and 3) when there is no
possibility of confusion and AnyW is used to express
that all the types of compartment can be concerned.
Concerning the optional components of a rule, a
specificity of our language is the neighbourhood pre-
conditions (PREC) that give the possibility to make
explicit some conditions to the occurrence of a re-
action. They consists in a list of pairs of compart-
ment variables to signify that instances of these vari-
ables must have a direct neighbourhood. Kinetic pa-
rameters (KParam) are inspired of BIOCHAM. They
consist in some arithmetic terms ranging from con-
stant values to more complex expressions based on
the identifiers given in the KPS section and the con-
centrations of molecules.
3.4 The Target Model
Once an exchange graph and a generic model have
been designed, the next step consists in instantiating
the generic reaction rules with topological informa-
tions given by the exchange graph in order to build
a concrete model. This last one is defined by all
valid instances of generic rules, obtained by replacing
compartment variables by compartments of the ex-
change graph, provided that conditions on compart-
ment types and compartments neighbourhoods are
satisfied in the exchange graph. For an exchange
graph EG = (V
EG
, E
EG
), a concrete rule cr is valid
if the three following conditions are met where SL
cr
and SR
cr
represent the sets of compartments appear-
ing in respectively the left hand and the right hand of
cr: i) the neighbourhood preconditions associated to
the rule are verified ; ii) the sub-graph resulting from
the restriction of EG to SL
rc
is connex or reduced to
one vertex ; and iii) each element SR
cr
is an element
of SR
rc
or has a direct neighbour in SL
cr
. For exam-
ple, the instantiation of
gr
1
: [M1 + M2]@Cyt => [M1]@Cyt&[M2]@Nuc.
relatively to GE
S
1
(Figure 4) gives two concrete
rules cr
11
and cr
12
, instances of gr
1
in which the
couple (Cyt, Nuc) has been respectively replaced by
(C1, N1) and (C2, N2).
The concrete model can then be exploited for simu-
lation and/or analysis issues, by using features pro-
vided by BIOCHAM and/or Pathway Logic. It suf-
fices to translate the concrete model in the concrete
syntax used by the targeted tool.
4 CASE STUDIES
In this section, we apply our approach on two pro-
cesses that have already been modelled in the lan-
guages of BIOCHAM or PL. The application pro-
ceeds in three steps: considering the model from
BIOCHAM or PL, we firstly infer from it information
about the involved localizations and the modelled re-
actions. This information is used to elaborate the ex-
change graph and the generic model to be used in the
coupling process. Then, we proceed to the coupling
and finally, the resulted concrete model is translated
in BIOCHAM and PL and the obtained target models
can be compared to the initial one. The models we
have chosen to consider are the model of the simpli-
fied cell cycle of Tyson (Model
1
) and the the model of
the delta-notch signalization pathway (Model
2
). They
are issued from BIOCHAM.
From Model
1
that contains ten reaction rules and no
mention of localization, we infer that the process is
concerned by ten different reactions, occurring in the
system seen as a whole. This information induce a
one-vertex (namely E of type Env) exchange graph
and a generic model whose rules are the transcription
in generic rules language of the ten reactions.
Model
2
contains one hundred and forty-four (abbrevi-
ated in seventy-two) reaction rules implicating thirty
six cells arranged in a 6x6 matrix, each cell being
identified by using its line and column numbers (C
11
,
C
12
... C
66
). Each of the rules of the model concerns
the synthesis or the degradation of one the two in-
volved molecules, Delta and Notch. That allows that
the system is about four biochemical reactions occur-
ring in a population cells of of thirty six (36) the same
A Proposal for a Language Combining Biochemical Rules and Topological Structure for Systems Biology
315

type. Conditions attached to the rules show that the
synthesis of Notch is juxtacrine (the occurrence of the
reaction in a given compartment depends on the con-
tents of the juxtaposed compartments) When adopting
the same topology as BIOCHAM, we can elaborate
the bio-geometric model of Figure 8. The exchange
graph extracted from this geometric model exactly
conforms what was expected : Except the neighbour-
ing with E, all the cells have between two and four
neighbours. The generic model contains four rules
corresponding to the transcription of the four reac-
tions in the generic rules language.
Figure 8: Bio-geometric model of a system consisting in a
population of thirty six (36) cells arranged in a 6x6 matrix.
The coupling step in the case of Model
1
consists in
just eliminating the information about localization,
because it’s the default compartment Env. The con-
crete model is then equivalent to the generic one when
considering the number of rules. The target model,
obtained by syntax adaptation, conserves the seman-
tic and have the same number of rules.
The coupling of the generic model and the exchange
graph issued from the decoupling of Model
2
gener-
ates a concrete model with one hundred and forty four
(144) rules: four rules by cell. That means that each
generic rule has been instanciated for each cell, re-
gardless of the number of the neighbouring cells it
has. latter. The translation of the concrete model in
BIOCHAM conserves the semantic.
The presented approach intends to model processes in
BIOCHAM and PL languages with the guarantee that
the obtained models are respectful from the topology
of the system. Such models can be directly defined
in the targeted tools considering an implicit topology.
However, applying the proposed approach presents
some advantages: It allows the modeller to have a
directory of generic models and another of topology
specifications. As a consequence it is possible to him
to easily study a set of biochemical reactions mod-
elled as a set of generic rules relatively to different
topologies and inversely: any combination of an ex-
change graph and a generic model defines a model.
The generic rules, by characterizing the types of com-
partments involved, contribute to inform a little more
on the role of the different types of compartment. An-
other advantage of the approach is the provided capa-
bility to study the same model thought the two target
tools: even if these tools presents a lot of similari-
ties, the analysis they carry out are not exactly the
same. Different analysis results may be interesting.
Naturally, because generic models are concise, they
are easier to edit.
Based on the cases presented above, we can say that
these interests are diversely relevant according to the
considered topology. For a system with just one com-
partment like Model
1
, just the second point is relevant
because the topology cannot change in any way. For
cases like Model
2
which concerns a topology char-
acterized by an important number of compartments
of the same type, and reactions largely depending on
neighbourhoods, all the interests are relevant.
5 CONCLUSION
In this paper, we have introduced an approach to mod-
elling biological processes by decoupling the topol-
ogy of the system and the biochemical reactions oc-
curring in the system. The topology is abstracted by
an exchange graph that contains the relevant informa-
tion on each compartment of the system : its name,
its type, its content and its neighbourhoods. The re-
actions in turn are modelled as rules characterizing
the types of compartments involved. Our approach
can be considered generic at tow levels. First of all,
it is generic with regard to the topology: a generic
model can be declined according to several graphs
of exchange, giving rise to as many concrete models.
These concrete models can then be studied through
different analysis tools (two in our case: BIOCHAM
and PL): this constitutes the second aspect of generic-
ity of our approach.
The use of geometric modelling to represent topolog-
ical pieces of information is a second trait of original-
ity of our approach. This allows us to design generic
models that are at least as concise as resulting con-
crete models and that are consistent models from a
topological point of view.
REFERENCES
Bartocci, E. and Lio, P. (2016). Computational modeling,
formal analysis, and tools for systems biology. PLOS
Computational Biology, 12(1):1–22.
Blinov, M. L., Schaff, J. C., Vasilescu, D., Moraru, I. I.,
Bloom, J. E., and Loew, L. M. (2017). Compartmen-
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
316

tal and spatial rule-based modelling with virtual cell.
Biophysical Journal, 113(8):1365–1372.
Boutillier, P., Maasha, M., Li, X., Medina-Abarca, H. F.,
Krivine, J., Feret, J., Cristescu, I., Forbes, A. G., and
Fontana, W. (2018). The kappa platform for rule-
based modeling. Bioinformatics.
Calzone, L., Chabrier-Rivier, N., Fages, F., and Soliman, S.
(2005). Apprentissage de règles de réactions biochim-
iques à partir de propriétés en logique temporelle. In
Guy Perrière, A. G. e. C. G., editor, Actes de JO-
BIM’05, pages 183–192, Lyon.
Cardelli, L. (2004). Brane calculi. In Computational
Methods in Systems Biology, International Confer-
ence, CMSB 2004, volume 3082 of Lecture Notes in
Computer Science, pages 257–278. Springer.
Chen, C.-C. and Crilly, N. (2016). Describing complex de-
sign practices with a cross-domain framework : learn-
ing from synthetic biology and swarm robotics. Res
Eng Design, 27:291–305. 10.1007/s00163-016-0219-
2.
Clavel, M., Duran, F., Eker, S., Lincoln, P., Marti-Oliet,
N., Meseguer, J., and Quesada, J. (2000). Towards
Maude 2.0. In 3rd International Workshop on Rewrit-
ing Logic and its Applications (WRLA’00), volume 36
of Electronic Notes in Theoretical Computer Science.
Elsevier.
Danos, V. and Laneve, C. (2004). Formal molecular biol-
ogy. Theoretical Computer Sciences, 325(1):69–110.
Dill, D. L., Merrill, K. A., Pamela, G., Talcott, C. L., Lader-
oute, K., and Lincoln, P. (2005). The pathalyzer:
A tool for analysis of signal transduction pathways.
In Proceedings of the First Annual Recomb Satellite
Workshop on Systems Biology, pages 11–22.
Eker, S., Knapp, M., Laderoute, K., Lincoln, P., and Tal-
cott, C. (2002). Pathway Logic: Executable models of
biological networks. In Proceedings of Fourth Inter-
national Workshop on Rewriting Logic and Its Appli-
cations (WRLA).
Faeder, J. R., Blinov, M. L., Goldstein, B., and Hlavacek,
W. S. (2005). Rule-based modeling of biochemical
networks. Complexity, 10(4):22–41.
Faeder, J. R., Blinov, M. L., and Hlavacek, W. S. (2009).
Rule-based modeling of biochemical systems with
bionetgen. Systems Biology, 10(4).
Fages, F., Gay, S., and Soliman, S. (2015). Inferring re-
action systems from ordinary differential equations.
Theoretical Computer Science, 599:64 – 78. Ad-
vances in Computational Methods in Systems Biol-
ogy.
Fages, F. and Soliman, S. (2008). Formal cell biology
in biocham. In Formal Methods for Computational
Systems Biology, 8th International School on Formal
Methods for the Design of Computer, Communication,
and Software Systems, SFM 2008, Bertinoro, Italy,
Advanced Lectures, volume 5016 of Lecture Notes in
Computer Science, pages 54–80. Springer.
Fages, F., Soliman, S., and Chabrier-Rivier, N. (2004).
Modelling and querying interaction networks in the
biochemical abstract machine biocham.
Giannakis, K. and Andronikos, T. (2017). Membrane au-
tomata for modeling biomolecular processes. Natural
Computing, 16(1):151–163.
Harris, L. A., Hogg, J. S., and Faeder, J. R. (2009). Com-
portmental rule-based modeling of biochemical sys-
tems. In Proceedings of the 2009 Winter Simulation
Conference. M. D. Rossetti, R. R. Hill, B. Johansson,
A. Dunkin, and R. G; Ingalls, eds.
Hlavacek, W. S., Faeder, J. R., Blinov, M. L., Posner, R. G.,
Hucka, M., and Fontana, W. (2006). Rules for mod-
eling signal-transduction systems. Sciences STKE,
344(6).
Li, C., Donizelli, M., Rodriguez, N., Dharuri, H., Endler,
L., Chelliah, V., Li, L., He, E., Henry, A., Stefan, M.,
Snoep, J., Hucka, M., Novère, N. L., and Laibe, C.
(2010). Biomodels database: An enhanced, curated
and annotated resource for published quantitative ki-
netic models. BMC Syst Biol, 4(92).
Ma’ayan, A. (2017). complex systems biology. J. R. Soc
Interface, 14(20170391).
Machado, D., Costa, R. S., Rocha, M., Ferreira, E. C.,
Tidor, B., and Rocha, I. (2011). Modeling formalisms
in systems biology. AMB Express a SpringerOpen
Journal.
Pedersen, M., P.hillips, A., and Plotkin, G. D. (2015).
A high-level language for rule-based modelling.
PLoSONE10(6):e0114296.doi:10.1371/journal.
pone.0114296.
Regev, A., Panina, E. M., Silverman, W., Cardelli, L., and
Shapiro, E. Y. (2004). Bioambients: an abstraction
for biological compartments. Theor. Comput. Sci.,
325(1):141–167.
Riesco, A., Santos-Buitrago, B., Rivas, J. D. L., Knapp, M.,
Santos-Garcia, G., and Talcott, C. (2017). Epidermal
growth factor signaling towards proliferation: Model-
ing and logic inference using forward and backward
search. BioMed Research International, 2017:11.
https://doi.org/10.1155/2017/1809513.
Suderman, R. and Hlavacek, W. S. (2017). Truml: A
translator for rule-based modeling languages. In Pro-
ceedings of the 8th ACM International Conference
on Bioinformatics, Computational Biology,and Health
Informatics, ACM-BCB ’17, pages 372–377, New
York, NY, USA. ACM.
Talcott, C. (2016). The pathway logic formal modeling sys-
tem: Diverse views of a formal representation of sig-
nal transduction. In Workshop on Formal Methods in
Bioinformatics and Biomedicine.
Talcott, C. and Dill, D. L. (2005). The pathway logic assis-
tant. In Proceedings of the Workshop Computational
Methods in Systems Biology (CMSB, G. Plotkin, Ed.,
2005, pages 228–239.
Vidil, F., Damiand, G., Dexet-Guiard, M., Guiard, N.,
Ledoux, F., Fousse, A., Fradin, D., Liang, Y., Men-
eveaux, D., and Bertrand, Y. (2002). Moka: 3d topo-
logical modeler.
A Proposal for a Language Combining Biochemical Rules and Topological Structure for Systems Biology
317
