
Constraint Maximal Inter-molecular Helix Lengths within RNA-RNA
Interaction Prediction Improves Bacterial sRNA Target Prediction
Rick Gelhausen
1
, Sebastian Will
2
, Ivo L. Hofacker
2
, Rolf Backofen
1,3
and Martin Raden
1
1
Bioinformatics Group, University of Freiburg, Georges-Koehler-Allee 106, 79110 Freiburg, Germany
2
Institute for Theoretical Chemistry, University of Vienna, Waehringer Strasse 17, 1090 Wien, Austria
3
Centre for Biological Signalling Studies (BIOSS), University of Freiburg, Schaenzlestr. 18, 79104 Freiburg, Germany
Keywords:
RNA-RNA Interaction Prediction, Steric Constraints, Constrained Helix Length, Canonical Helix, Seed.
Abstract:
Efficient computational tools for the identification of putative target RNAs regulated by prokaryotic sRNAs
rely on thermodynamic models of RNA secondary structures. While they typically predict RNA–RNA in-
teraction complexes accurately, they yield many highly-ranked false positives in target screens. One obvious
source of this low specificity appears to be the disability of current secondary-structure-based models to reflect
steric constraints, which nevertheless govern the kinetic formation of RNA–RNA interactions. For example,
often—even thermodynamically favorable—extensions of short initial kissing hairpin interactions are kineti-
cally prohibited, since this would require unwinding of intra-molecular helices as well as sterically impossible
bending of the interaction helix. In consequence, the efficient prediction methods, which do not consider such
effects, predict over-long helices. To increase the prediction accuracy, we devise a dynamic programming
algorithm that length-restricts the runs of consecutive inter-molecular base pairs (perfect canonical stackings),
which we hypothesize to implicitely model the steric and kinetic effects. The novel method is implemented by
extending the state-of-the-art tool INTARNA. Our comprehensive bacterial sRNA target prediction benchmark
demonstrates significant improvements of the prediction accuracy and enables 3-4 times faster computations.
These results indicate—supporting our hypothesis—that length-limitations on inter-molecular subhelices in-
crease the accuracy of interaction prediction models compared to the current state-of-the-art approach.
1 INTRODUCTION
Small RNAs (sRNAs) are central regulators in
prokaryotic cells (Storz et al., 2011). For instance,
they can trigger mRNA decay (Lalaouna et al., 2013)
or modulate translation (Hoe et al., 2013) via di-
rect inter-molecular base pairing. Different mech-
anisms are known (detailed e.g. in (Nitzan et al.,
2017)) like the blocking of the ribosomal binding site
causing translation inhibition or the (de-)stabilization
of mRNAs by covering (or providing) binding sites
of RNAases. The sRNA–RNA interactions typically
contain a small nearly perfect subinteraction of about
7 base pairs (known as seed region) (K
¨
unne et al.,
2014) and have been shown to be located at acces-
sible regions that are mainly unpaired (Richter and
Backofen, 2012). Thus, beside general RNA–RNA
interaction prediction approaches (reviewed e.g. in
(Wright et al., 2018)), dedicated prediction tools like
INTARNA (Busch et al., 2008) or RNAPREDATOR
(Eggenhofer et al., 2011) have been developed and
applied (Li et al., 2012). Recently, fast heuristics
for genome-wide screens have been implemented,
e.g. RIBLAST (Fukunaga and Hamada, 2017) and
RISEARCH2 (Alkan et al., 2017).
For elucidating the regulatory network of sRNAs,
target prediction is applied (Backofen et al., 2014)
to guide experimental validation. While the essen-
tial bioinformatics machinery for this task is avail-
able (Backofen et al., 2017), computational meth-
ods still predict a high number of false positive tar-
gets. The latter can be reduced when individual tar-
get predictions of homologous sequences (Lott et al.,
2018) are combined in comparative approaches like
COPRARNA (Wright et al., 2014). Unfortunately,
this technique is only applicable for the identification
of evolutionary conserved targets. Another option is
to incorporate experimental structure probing data to
emend the RNAs’ accessibility information (Miladi
et al., 2019). Integrating probing data, which can be
obtained from high-throughput experiments (Choud-
hary et al., 2017), can significantly alleviate the prob-
Gelhausen, R., Will, S., Hofacker, I., Backofen, R. and Raden, M.
Constraint Maximal Inter-molecular Helix Lengths within RNA-RNA Interaction Prediction Improves Bacterial sRNA Target Prediction.
DOI: 10.5220/0007689701310140
In Proceedings of the 12th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2019), pages 131-140
ISBN: 978-989-758-353-7
Copyright
c
2019 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
131

lem of inaccurate accessibility prediction. However, it
does not touch—and is even orthogonal to—the here
discussed issues of target prediction.
In this work, we study means to efficiently im-
prove sRNA target prediction by restricting the ad-
missible interaction patterns. This is hypothesized to
incorporate steric and kinetic aspects going beyond
the thermodynamic secondary structure-based mod-
els. Specifically, our method is motivated by the ob-
servation that interacting sites of sRNAs are either
not enclosed by any base pairing (exterior) or located
within loop regions. For loop regions, the forma-
tion of (long) inter-molecular helices (i.e. the entan-
gling of the RNA molecules) requires the ‘unwinding’
of intra-molecular helices, which imposes additional
constraints on the substantial steric rearrangements
(rotating large parts of the molecules through space)
while the interaction grows. Consequently, the forma-
tion of long inter-molecular duplexes seems to be pro-
hibited, even if it would be expected in the currently
used thermodynamic models due to high hybridiza-
tion stability and sufficient accessibility. This well-
known phenomenon has been studied in the context
of other loop-initiated RNA–RNA interactions (Kolb
et al., 2000; Brunel et al., 2002).
Concretely, we test whether interaction predic-
tion can be improved by explicitly constraining the
maximal length of inter-molecular helices, which—
as we conjecture—indirectly considers steric and ki-
netic constraints. While preserving tractability, limit-
ing this length ensures that long helices must be in-
terrupted by interior loops—which is thought to re-
lax the ‘winding tension’. We provide efficient dy-
namic programming algorithms both for exact as well
as heuristic interaction prediction, incorporating the
new helix-length constraint (in addition to the well-
established seed constraint of previous approaches).
The approach is incorporated into INTARNA (Mann
et al., 2017), a state-of-the-art RNA–RNA interaction
prediction tool (Umu and Gardner, 2017). Finally,
we assess the effect of the helix-length constraints on
a large prokaryotic sRNA target prediction data set
extending (Wright et al., 2013). In this benchmark,
the helix length limitation reduces the overall runtime
and, supporting our conjecture, improves the predic-
tion quality.
2 METHODS
In the following, we will first present the recursions
used by the current state-of-the-art prediction ap-
proaches like RNAUP (M
¨
uckstein et al., 2006) or IN-
TARNA (Mann et al., 2017). Subsequently, we in-
troduce the new recursions for helix-length restricted
prediction. First, all recursions are given for ex-
haustive/optimal interaction prediction, followed with
a discussion how they can be turned into efficient
heuristic variants. To ease readability, we provide
graphical recursion depictions and provide respective
formulas in the Appendix.
2.1 Accessibility-based Interaction
Prediction
Given two RNAs S
1
,S
2
of length n,m, resp., we want
to find the interaction sites i..k ∈ [1,n] of S
1
and
j..l ∈ [1, m] of S
2
that minimize the interaction en-
ergy E(i, j,k, l). That is, we are interested in the most
stable interaction of an sRNA with a given putative
target. This interaction energy can then be used for
target ranking and the selection of the most promis-
ing candidates.
The interaction sites are considered free of intra-
molecular base pairs and can only form inter-
molecular base pairs. Two positions of the RNAs
can form a base pair if the respective nucleotides
are complementary (i.e. AU, GC, or GU). We con-
sider only sites where the boundaries are forming two
inter-molecular base pairs (i, j),(k,l). No two inter-
molecular base pairs (x, y),(x
0
,y
0
) ∈ [1, n] × [1, m] are
allowed to be crossing, i.e. it holds x ≤ x
0
↔ y ≤
y
0
, nor allowed to share a position within the same
RNA. Following the Nearest Neighbor energy model
(Tinoco Jr et al., 1973), the hybridization or du-
plex formation energy of a site is thus given by
the sum of the loop energies (Turner and Mathews,
2010) defined by consecutive base pairs. Here, we
distinguish between directly neighbored base pairs,
scored by E
S
terms, and neighbored base pairs that
enclose unpaired positions, evaluated by E
IL
terms.
The hybridization energy also contains a general en-
ergy penalty term E
init
that, to some extent, reflects
the probability of interaction initiation. The optimal
(minimal) hybridization energy among all possible in-
teractions of the sites is given by H(i, j,k, l). The en-
ergy penalty ED needed to break all intra-molecular
base pairs within the individual sites is used to incor-
porate the sites’ accessibility for interaction forma-
tion. The overall energy of a site is thus given by
E(i, j,k, l) = H(i, j, k,l) + ED(i..k)+ ED( j..l). (1)
All energy terms presented in the following are given
in kcal/mol unit and are computed using the Vienna
RNA package (Lorenz et al., 2011) version 2.4.4. For
simplicity, we exclude dangling-end and helix-end
contributions within Eq. 1. For formalisms, we refer
to the detailed introduction provided in (Raden et al.,
2018).
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
132
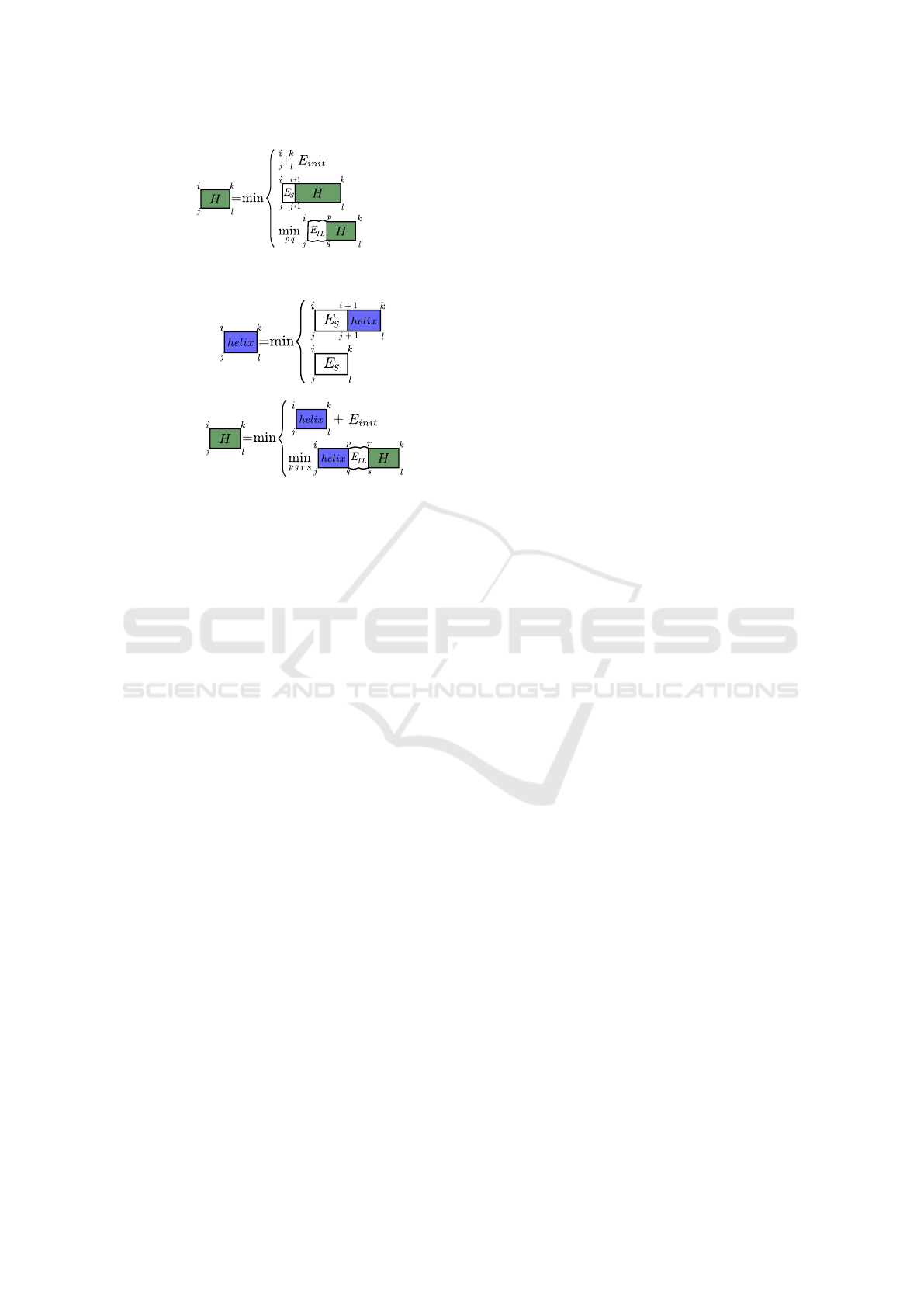
=
=
Figure 1: Sketch of the state-of-the-art recursion to compute
the optimal interaction energy without further constraints.
Figure 2: Recursion depictions to compute canonical helix
energies helix (top) using energy terms E
S
for stacked base
pairs and the optimal energy H (bottom) for a given inter-
action site using the energy terms E
IL
for interior loops.
ED terms can be efficiently computed via dy-
namic programming (Bernhart et al., 2011). This
leaves the computation of the optimal interaction en-
ergy H, also accessible via dynamic programming
(M
¨
uckstein et al., 2006). Figure 1 visualizes the cen-
tral recursion that either scores an initial base pair
(E
init
) or extends a shorter optimal interaction with
a stacked base pair (E
S
) or an interior loop contribu-
tion (E
IL
). All individual energy contributions E
init
,
E
S
and E
IL
are +∞ if the respective boundary indices
are non-complementary, i.e. can not form a base pair.
Note, interior loop sizes (p-r and q-s) are typically re-
stricted to a fixed maximal length w n, m, which re-
sults in a runtime complexity of O(n
2
m
2
). A heuristic
variant of this recursion available in INTARNA with
O(nm) runtime was introduced in (Busch et al., 2008).
The base pairs of an optimal interaction with energy
H(i, j,k, l) can be obtained via traceback if of interest.
2.2 Helix-length Restricted Prediction
In order to restrict the length of inter-molecular he-
lices to a predefined constant c
B
≥ 2, referred to as he-
lix length, we decompose the prediction process into
two steps: (a) the energy pre-computation of possi-
ble helices composed of at most c
B
base pairs, and (b)
their assembly in order to find the optimal interaction
energy for a given site.
For simplicity, we first consider canonical he-
lices, i.e. perfect helices composed of stacked base
pairs only. Adaptions to non-canonical helices con-
taining small bulges and interior loops are discussed
in a subsequent section. Figure 2 shows the recur-
sion to compute the energy of canonical helices with
the left-/right-most inter-molecular base pair (i, j) <
(k, l), resp., stored in helix(i, j,k,l). The length con-
straint c
B
is ensured for canonical helices by set-
ting all entries to +∞ if the helix is too long, i.e.
max(k − i, l − j) ≥ c
B
. Note, for non-canonical he-
lices, helix(i, j,k,l) will contain the optimal energy
of any helix fulfilling the relaxed constraints.
Given this, the optimal hybridization energy
H(i, j,k, l) for the given interaction sites i..k and j..l
can be computed via the recursion depicted in Fig. 2.
That is, we either consider a full helix (if possible for
the given boundaries) or compose an interaction via
the addition of a new helix (on the left) to extend a
smaller optimal interaction. The composition inserts
an interior loop between the helix and the next inter-
action to ensure that no two helices are combined into
a longer one. Thus, the interior loop has to span at
least one unpaired position, i.e. (p − r ) + (q − s) > 2,
and is constrained in length as for the recursions dis-
cussed before. Since both helix length as well as in-
terior loop length are constrained by respective con-
stants c
B
and w, the overall runtime complexity is still
O(n
2
m
2
).
2.3 Enforcing Seed Constraints in
Helix-length Restricted Prediction
As already discussed, seed-constraints are a central
tool to reduce false positive sRNA target predictions
(Tjaden et al., 2006; Bouvier et al., 2008). Within IN-
TARNA, possible seed interactions and respective en-
ergies are efficiently computed via dynamic program-
ming analogously to the presented helix energy pre-
processing; please refer to (Busch et al., 2008) for de-
tails. In the following, the optimal energy for the seed
with left-/right-most base pairs (i, j),(k,l), resp., are
stored in seed(i, j, k, l).
In order to ensure that a reported interaction con-
tains a seed region, we follow the approach presented
in (Busch et al., 2008). Therein, a second dynamic
programming table H
S
is computed based on H that
provides the optimal energy for a site given that the
considered interaction contains a seed region. The op-
timal energy of a site with seed is then given by
E(i, j,k, l) = H
S
(i, j, k, l) + ED(i..k) + ED( j..l) (2)
replacing Eq. 1.
Since seeds are valid parts of helices, which are
the building blocks for our introduced H computation,
we use a second auxiliary matrix helix
S
that provides
the optimal helix energy given that the helix contains
Constraint Maximal Inter-molecular Helix Lengths within RNA-RNA Interaction Prediction Improves Bacterial sRNA Target Prediction
133

Figure 3: Depiction of the decomposition strategy for the
computation of the optimal energy of a helix containing a
seed helix
S
(top) and the best hybridization energy H
S
(bot-
tom) enforcing both the helix and seed constraint.
a seed. If the region contains no valid seed or this
would lead to too many base pairs, the energy is set
to +∞. Figure 3 depicts the recursion in order to fill
helix
S
based on the already introduced helix informa-
tion that is combined with the seed energy. To this
end, all possible locations of a seed combined with
flanking helices are evaluated. Due to the indepen-
dence of the seed and helix constraints, it is possible
to allow unpaired bases in the seed, even when not
allowing unpaired bases in the helix constraints and
vice versa.
Given this, the optimal hybridization energy H
S
for a given site containing a seed and only helices
with at most c
B
base pairs, can be computed using
a recursion as depicted in Fig. 3. That is, either (i)
the site can be filled with a single helix containing a
seed (plus accounting for interaction initiation), or (ii)
a helix-length-constrained interaction site is extended
with a seed-containing helix, or (iii) we extend an in-
teraction that contains already a seed with a helix that
is not constrained to contain a seed.
2.4 Enforcing a Minimal Helix Stability
Given our focus on helices, we can easily enforce ad-
ditional constraints on the helices that are considered
for interaction composition. As the first step, we in-
troduce a minimal stability notion via an upper hy-
bridization energy bound E
helix
max
for individual helices.
Since energy is inversely related to stability, our ap-
proach will produce interaction patterns of stable sub-
helices connected by interior loop regions.
The energy threshold can be easily incorporated
into the presented recursions by extending the com-
putation of H and H
S
from Fig. 2 and 3, resp., with
side conditions. That is, entries from helix or helix
S
are only considered, if the respective energy value is
below the given threshold E
helix
max
.
-
1
Figure 4: Recursion depiction to compute the optimal hy-
bridization energy helix
U
(i, j,k,l,B) for non-canonical he-
lices with exactly B base pairs and interior loops containing
at most c
U
unpaired bases, i.e. (p − i) + (q − j) ≤ c
U
+ 2
and analogously for the second case.
2.5 Consideration of Non-canonical
Helices
So far, we only considered canonical helices for the
computation of helix. While this models the most sta-
ble helices that can be formed, minor variance of this
ideal, i.e. allowing for bulges or interior loops span-
ning only single or very few unpaired bases, will still
resemble a stable helix. But considering stable helices
only (using E
helix
max
) would likely exclude such helices
if the canonical subhelices are too short. Thus, we
next discuss how the hybridization energy helix for
helices including minor bulges of at most c
U
unpaired
bases can be computed. We consider an interior loop
as minor if c
U
≤ 2.
To this end, we introduce the auxiliary matrix
helix
U
(i, j, k, l,B) that provides the optimal helix hy-
bridization energy for the given site boundaries and
the number of base pairs B while allowing minor
bulges of size c
U
. Figure 4 depicts the respective re-
cursion. Note, the boundaries p,q considered for inte-
rior loops are constrained to (p−i)+(q− j) ≤ c
U
+2.
The optimal helix hybridization energy helix(i, j,k, l)
is thus given by helix
U
(i, j, k, l,c
B
). Note, enforcing
the helices to be stable (via E
helix
max
) will without further
constraints exclude helices composed of bulges only.
In addition to the altered helix computation, we
also have to ensure that the helices assembled within
the H and H
S
computation are spaced by interior
loops exceeding c
U
. That is, it holds for Fig. 2 and 3
that (r − p) + (s − q) > c
U
+ 2. Note that setting
c
U
= 0 will provide the same results as if using canon-
ical helices only.
2.6 Heuristic Helix-length Restricted
Prediction
Due to the high time and space complexity of the ex-
act approach, we implemented heuristic variants of
the recursions following the ideas from (Busch et al.,
2008) introduced for INTARNA. That is, instead of
considering all interaction ranges for a given left-most
base pair (i, j), only the optimal right boundary (k,l)
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
134

together with the respective hybridization energy is
stored in H and H
S
. Thus, for a given left-most base
pair (i, j), the recursions from above are not confined
to a specific right k, l bound but use the right end of
the respective optimal recursion case. Please refer to
(Busch et al., 2008) for further details. This heuris-
tic reduces the space and time complexity to O(nm),
provides almost the same prediction quality (Umu and
Gardner, 2017), and makes the approach feasible for
the needed large-scale target screens also discussed in
the Result section.
Here, we apply this strategy not only to H and H
S
but also to the helix and helix
S
matrices. That is, we
only memorize the best helix energy (and right bound-
ary) for each left-most helix base pair (i, j). Note,
both matrices have to be computed using small aux-
iliary matrices that replace the respective recursions.
Note further, the computation of H and H
S
becomes
more simple, since we do not consider different helix
lengths (via p and q) but only use the right-most base
pair of the best helix with left-most base pair (i, j).
3 RESULTS
To evaluate our introduced predictors concerning their
sRNA target prediction performance, we introduce
the manually curated benchmark data used subse-
quently.
3.1 Data Set for sRNA Target
Prediction Benchmark
We investigate whether a restriction on inter-
molecular helix lengths could improve the overall pre-
diction accuracy of INTARNA. To this end, we cre-
ated an sRNA target prediction benchmark extending
the ideas and data from (Wright et al., 2013). The
whole benchmark data set including respective scripts
is available at:
https://github.com/BackofenLab/IntaRNA-benchmark.
The benchmark consists of a large set of bacterial
sRNA queries and potential target sequences. We re-
strict our analysis to sRNA regulation based on the
blocking of the ribosomal binding site (see (Nitzan
et al., 2017) for a discussion). Thus, the targets
are genomic sub-regions around the start codon of
the respective mRNA including 200 nucleotides up-
stream and 100 nucleotides downstream, since many
sRNAs that regulate translation bind their target in
a region around the start codon. Sequences are ex-
tracted from the GenBank database of the National
Centre for Biotechnology Information (NCBI) (Ben-
son et al., 2008). The dataset comprises 4,319 target
regions from the E.coli genome (GenBank accession
number NC 000913) and 4,552 target regions from
the Salmonella typhimurium genome (NC 003197).
The query data set consists of 15 sRNAs from E. coli
and 15 from Salmonella, which have been shown ex-
perimentally to act as post-transcriptional regulators
by base-pairing to at least one of the target mRNAs.
To evaluate the performance, we follow the ap-
proach from (Tjaden et al., 2006). To this end, we
extracted 149 sRNA-mRNA pairs from the literature
that have been experimentally verified to interact.
Within the benchmark, we test how well these veri-
fied pairs can be separated from all possible sRNA-
mRNAs pairs. That is, we predict the optimal interac-
tion energy E for each of the 15 sRNAs in E.coli with
any of the 4,319 putative target regions. The same is
done for the Salmonella data set. As a result, there are
in total 133,065 potential sRNA-target interactions
and respective interaction energy estimates. From
these, only the mentioned 149 query-target pairs are
supported, leaving 132,916 unsupported pairs. Fi-
nally, we test whether interactions of the verified pairs
have lower optimal energy estimates compared to the
unsupported interactions. In other words, we evalu-
ate the ranks of the supported pairs within the energy
score distribution over all putative targets. That is, the
more verified interactions are predicted with low rank,
the more precise is the target prediction approach. A
detailed description of the technical part is available
in the Appendix.
3.2 Helix-length Constraints Enable
Faster Predictions and Improve
Prediction Quality
As reference and ”gold standard” for the evaluation of
our helix-length restricted approach, we use the pre-
diction performance of INTARNA version 2.2.0 us-
ing default values (i.e. heuristic predictions including
seeds) on the introduced benchmark data set using a
seed of length 7 for all predictions. In the following,
we refer to this version with ”original”.
Effect of Maximal Helix Length
We have extended INTARNA with implementations
of our heuristic recursions, which enables clean com-
parisons for both prediction quality as well as space
and runtime requirement of the computations. Fig-
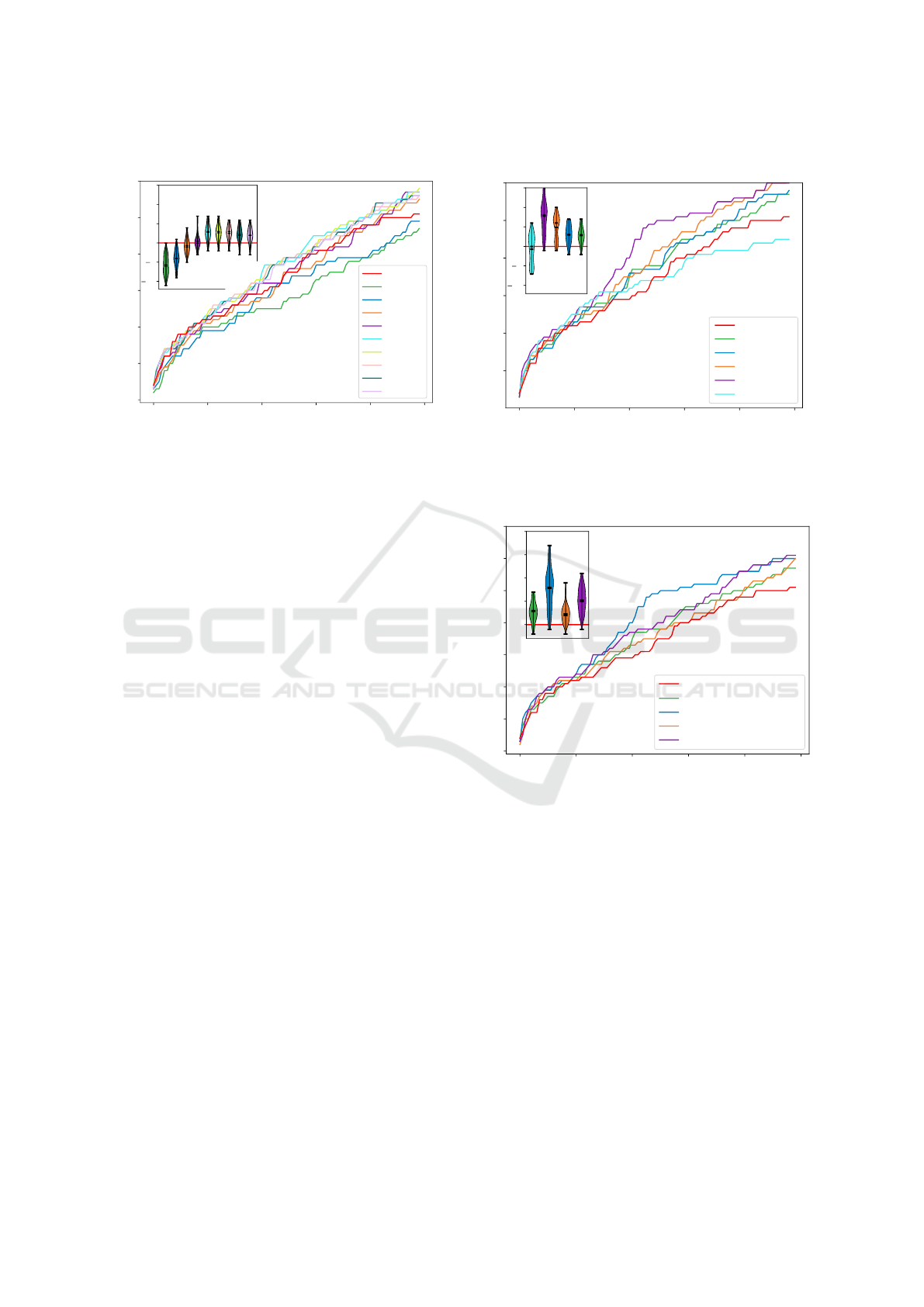
ure 5 compares the results for different maximal helix
length values c
B
with the original predictions. The
Constraint Maximal Inter-molecular Helix Lengths within RNA-RNA Interaction Prediction Improves Bacterial sRNA Target Prediction
135
# top-ranked targets per sRNA
10
5
0
5
10
15
0 20 40 60 80 100
0
10
20
30
40
50
60
sum of verified sRNA-target pairs
original
deviation
Performance for length-limited canonical stacks
containing a seed
c
B
=7
c
B
=8
c
B
=9
c
B
=10
c
B
=11
c
B
=12
c
B
=13
c
B
=14
c
B
=15
Figure 5: Effect of different maximal canonical helix
lengths c
B
in terms of the recovery of verified sRNA-target
pairs among the top ranked predictions compared to the
original INTARNA results (red curve). The inset shows the
differences to the original results (red).
curves visualize the total number of verified sRNA-
target pairs within the respective top-ranked predic-
tions for each sRNA. That is the higher the curve the
better the recovery rate of the verified targets. To ease
comparisons, the inset shows the difference between
the results of the original version (red curve) and the
respective helix-length constrained predictions.
For low c
B
values, we observe a reduced predic-
tion accuracy compared to the original recursions. In
contrast, maximal helix lengths of 11-13 show much
improved recovery rates and provide the overall best
results. For c
B
= 11, we observe the highest recov-
ery improvement of additional 2.7 verified targets on
average. Higher c
B
values have a lower prediction ac-
curacy that will eventually converge towards the un-
constrained original prediction results. These obser-
vations apply to all tested variants.
In addition to the higher prediction accuracy, the
constrained version is about 3-4 times faster com-
pared to the original version, while maintaining the
same memory consumption.
Effect of Minimal Helix Stability
Next, we investigate whether the restriction to stable
helices can further improve the prediction quality. To
this end, we fixed the maximal helix length to c
B
= 11,
given the results from above, and tested different he-
lix hybridization energy thresholds E
helix
max
. Figure 6
summarizes the results. E
helix
max
= −7.5 shows best
performance with a recovery improvement of about
8 verified targets on average, which is about three-
times higher compared to c
B
= 11 results without sta-
10
5
0
5
10
15
0 20 40 60 80 100
10
20
30
40
50
60
maxE=0
maxE=-2.5
maxE=-5
maxE=-7.5
maxE=-10
original
deviation
Performance for stable length-limited canonical
stacks containing a seed
# top-ranked targets per sRNA
sum of verified sRNA-target pairs
Figure 6: Effect of different maximal helix energies E
helix
max
(maxE in the plot) on the prediction performance (maximal
canonical helix length c
B
= 11) analogously to Fig. 5.
0 20 40 60 80 100
0
10
20
30
40
50
60
70
can. c
B
=11
can. c
B
=11 maxE=-7.5
c
U
=2 c
B
=13
c
U
=2 c
B
=13 maxE=-7.5
original
Best performing parameterizations
0
5
10
15
20
deviation
# top-ranked targets per sRNA
sum of verified sRNA-target pairs
Figure 7: Overview of the best prediction performance for
canonical (can.) (maximal canonical helix length c
B
=
11) and non-canonical (c
U
= 2) (maximal canonical helix
length c
B
= 13) and the overall best E
helix
max
= −7.5 for both
methods. Plot analogously to Fig. 5.
bility constraint (see above). Note, all tested E
helix
max
values above −7.5 provide improved prediction per-
formance. Too low thresholds (E
helix
max
= −10) exclude
too many helices to enable better target prediction.
Effect of Non-canonical Helices
When relaxing the helix definition to non-canonical
helices that are allowed to contain minor bulges or in-
terior loops with up to c
U
unpaired bases, the overall
prediction performance is not exceeding the canonical
helix variant. Here, best results are observed for c
U
=
2 and a maximal helix length c
B
= 13 while c
B
values
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
136

of 11-13 still show the best results (data not shown).
Figure 7 provides a comparison of the best parameter-
izations for the canonical and non-canonical approach
with and without E
helix
max
= −7.5 constraint. From this
it is obvious that a relaxation of the helix definition
does not improve the prediction quality compared to
stable canonical helices.
4 DISCUSSION & CONCLUSION
Predictions of helix-length constrained interactions
can be done with the same time and space complexity
as known for unconstrained RNA–RNA interaction
prediction. In fact, the helix-length constraint signifi-
cantly reduces the search space such that we observe
on average a 3-4 times faster target prediction for our
benchmark data set. The reduced runtime results from
the following: compared to the current state-of-the-art
recursion from Fig. 1, the helix-length constrained ap-
proach from Fig. 2 faces the same search space for the
interior loop sizes but appends full helices instead of
individual base pairs. This becomes even more strik-
ing for the heuristic variant, which does not consid-
ered all possible helix lengths but only the optimal
helix for the left-most base pair (i, j).
Furthermore, we observe enriched target predic-
tion accuracy measured in terms of increased recov-
ery rates of verified sRNA-target pairs known from
the literature. Maximal helix lengths c
B
of 11-13
base pairs show the best prediction quality, while
shorter drastically reduced the recovery rate. This
is mainly achieved by disregarding low-energy inter-
actions composed of many bulge and interior loops
(putative false positives) rather than altering the inter-
action details of the verified sRNA-target pairs. Re-
sults can be further improved when only stable he-
lices with an energy below a given threshold E
helix
max
are considered for prediction. For helices of a maxi-
mal length of 11-13 base pairs, an upper energy bound
of −7.5kcal/mol provides the best target prediction
performance. Notably, the consideration of a relaxed
helix definition allowing for small bulge or interior
loops does not significantly improve the results com-
pared to perfect canonical helices but has slightly
higher runtime requirements.
Our observations support our hypothesis that long
inter-molecular helices are less likely due to steric and
kinetic constraint of the interaction formation process.
That is, we think the ’decomposed helix interaction
model’, where short stable helices are interrupted by
flexible interior loops, a more realistic model com-
pared to unconstrained predictions.
One way to further improve the model would be
to confine helix-length constrained predictions to re-
gions mainly unpaired in loop regions while applying
the unconstrained approach for exterior unpaired re-
gions. This can be efficiently distinguished during ED
computation from the underlying partition functions
(M
¨
uckstein et al., 2006; Bernhart et al., 2011). An-
other planned direction is to apply further constraints
on the helices considered within the prediction. For
instance, we will further investigate the correlation of
helix base pair number c
B
and optimal upper energy
bounds E
helix
max
, since they are most likely linked by the
average stacking energy or similar terms.
Even though we exemplified our new approach by
extending INTARNA, the concept is a generic one.
We therefore expect also other RNA–RNA interaction
prediction methods to profit from a restriction of inter-
molecular helix lengths.
ACKNOWLEDGEMENTS
This work was supported by the German Research
Foundation (DFG) [BA2168/16-1] and the Austrian
Science Fund (FWF) [I 2874-N28].
REFERENCES
Alkan, F., Wenzel, A., Palasca, O., Kerpedjiev, P., Rude-
beck, A., Stadler, P. F., Hofacker, I. L., and Gorodkin,
J. (2017). RIsearch2: suffix array-based large-scale
prediction of RNARNA interactions and siRNA off-
targets. Nucleic Acids Research, 45(8):e60.
Backofen, R., Amman, F., Costa, F., Findeiss, S., Richter,
A. S., and Stadler, P. F. (2014). Bioinformatics of
prokaryotic RNAs. RNA Biol, 11(5).
Backofen, R., Engelhardt, J., Erxleben, A., Fallmann, J.,
Gr
¨
uning, B., Ohler, U., Rajewsky, N., and Stadler,
P. F. (2017). RNA-bioinformatics: Tools, services and
databases for the analysis of RNA-based regulation. J
Biotechnol, 261:76–84.
Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell,
J., and Wheeler, D. L. (2008). GenBank. Nucleic
Acids Res., 36(Database issue):25–30.
Bernhart, S. H., M
¨
uckstein, U., and Hofacker, I. L. (2011).
RNA accessibility in cubic time. Algorithms for
Molecular Biology, 6(1):3.
Bouvier, M., Sharma, C. M., Mika, F., Nierhaus, K. H., and
Vogel, J. (2008). Small RNA binding to 5’ mRNA
coding region inhibits translational initiation. Mol.
Cell, 32(6):827–837.
Brunel, C., Marquet, R., Romby, P., and Ehresmann, C.
(2002). RNA loop–loop interactions as dynamic func-
tional motifs. Biochimie, 84(9):925 – 944.
Busch, A., Richter, A. S., and Backofen, R. (2008). In-
taRNA: efficient prediction of bacterial sRNA tar-
gets incorporating target site accessibility and seed re-
gions. Bioinformatics, 24(24):2849–56.
Constraint Maximal Inter-molecular Helix Lengths within RNA-RNA Interaction Prediction Improves Bacterial sRNA Target Prediction
137

Choudhary, K., Deng, F., and Aviran, S. (2017). Compar-
ative and integrative analysis of RNA structural pro-
filing data: current practices and emerging questions.
Quantitative Biology, 5(1):3–24.
Eggenhofer, F., Tafer, H., Stadler, P. F., and Hofacker, I. L.
(2011). RNApredator: fast accessibility-based pre-
diction of sRNA targets. Nucleic Acids Res, 39(Web
Server issue):W149–54.
Fukunaga, T. and Hamada, M. (2017). RIblast: an ul-
trafast RNARNA interaction prediction system based
on a seed-and-extension approach. Bioinformatics,
33(17):2666–2674.
Hoe, C.-H., Raabe, C. A., Rozhdestvensky, T. S., and
Tang, T.-H. (2013). Bacterial sRNAs: Regulation in
stress. International Journal of Medical Microbiol-
ogy, 303(5):217 – 229.
Kolb, F. A., Malmgren, C., Westhof, E., Ehresmann, C.,
Ehresmann, B., Wagner, E. G., and Romby, P. (2000).
An unusual structure formed by antisense-target RNA
binding involves an extended kissing complex with
a four-way junction and a side-by-side helical align-
ment. RNA, 6(3):311–324.
K
¨
unne, T., Swarts, D. C., and Brouns, S. J. (2014). Plant-
ing the seed: target recognition of short guide RNAs.
Trends in Microbiology, 22(2):74 – 83.
Lalaouna, D., Simoneau-Roy, M., Lafontaine, D., and
Mass, E. (2013). Regulatory RNAs and target mRNA
decay in prokaryotes. Biochimica et Biophysica Acta
(BBA) - Gene Regulatory Mechanisms, 1829(6):742 –
747. RNA Decay Mechanisms.
Li, W., Ying, X., Lu, Q., and Chen, L. (2012). Predicting
sRNAs and their targets in bacteria. Genomics, Pro-
teomics & Bioinformatics, 10(5):276 – 284.
Lorenz, R., Bernhart, S. H., H
¨
oner zu Siederdissen, C.,
Tafer, H., Flamm, C., Stadler, P. F., and Hofacker,
I. L. (2011). ViennaRNA Package 2.0. Algorithms
for Molecular Biology, 6(1):26.
Lott, S. C., Sch
¨
afer, R. A., Mann, M., Backofen, R., Hess,
W. R., Voss, B., and Georg, J. (2018). GLASSgo -
automated and reliable detection of sRNA homologs
from a single input sequences. Frontiers in Genetics,
9:124.
Mann, M., Wright, P. R., and Backofen, R. (2017). In-
taRNA 2.0: enhanced and customizable prediction
of RNA-RNA interactions. Nucleic Acids Res.,
45(W1):W435–W439.
Miladi, M., Montaseri, S., Backofen, R., and Raden, M.
(2019). Integration of accessibility data from structure
probing into RNA-RNA interaction prediction. Bioin-
formatics, (epub ahead of print).
M
¨
uckstein, U., Tafer, H., Hackerm
¨
uller, J., Bernhart, S. H.,
Stadler, P. F., and Hofacker, I. L. (2006). Thermo-
dynamics of RNA–RNA binding. Bioinformatics,
22(10):1177–1182.
Nitzan, M., Rehani, R., and Margalit, H. (2017). Integra-
tion of bacterial small RNAs in regulatory networks.
Annual Review of Biophysics, 46(1):131–148.
Raden, M., Mohamed, M. M., Ali, S. M., and
Backofen, R. (2018). Interactive implementa-
tions of thermodynamics-based RNA structure and
RNA-RNA interaction prediction approaches for
example-driven teaching. PLOS Comput. Biol,
14(8):e1006341.
Richter, A. S. and Backofen, R. (2012). Accessibility
and conservation: General features of bacterial small
RNA-mRNA interactions? RNA Biol, 9(7):954–65.
Storz, G., Vogel, J., and Wassarman, K. (2011). Regula-
tion by small RNAs in bacteria: Expanding frontiers.
Molecular Cell, 43(6):880 – 891.
Tinoco Jr, I., Borer, P., Dengler, B., Levin, M., Uhlenbeck,
O., Crothers, D., and Bralla, J. (1973). Improved es-
timation of secondary structure in ribonucleic acids.
Nature New Biology, 246(150):40–41.
Tjaden, B., Goodwin, S. S., Opdyke, J. A., Guillier, M.,
Fu, D. X., Gottesman, S., and Storz, G. (2006). Tar-
get prediction for small, noncoding RNAs in bacteria.
Nucleic Acids Res., 34(9):2791–2802.
Turner, D. H. and Mathews, D. H. (2010). NNDB: the near-
est neighbor parameter database for predicting stabil-
ity of nucleic acid secondary structure. Nucleic Acids
Res, 38(Database issue):D280–2.
Umu, S. U. and Gardner, P. P. (2017). A comprehensive
benchmark of RNA-RNA interaction prediction tools
for all domains of life. Bioinformatics, 33(7):988–
996.
Wright, P. R., Georg, J., Mann, M., Sorescu, D. A., Richter,
A. S., Lott, S., Kleinkauf, R., Hess, W. R., and Back-
ofen, R. (2014). CopraRNA and IntaRNA: predicting
small RNA targets, networks and interaction domains.
Nucleic Acids Res., 42(Web Server issue):W119–23.
Wright, P. R., Mann, M., and Backofen, R. (2018). Struc-
ture and interaction prediction in prokaryotic RNA bi-
ology. Microbiol Spectrum, 6(2).
Wright, P. R., Richter, A. S., Papenfort, K., Mann, M.,
Vogel, J., Hess, W. R., Backofen, R., and Georg, J.
(2013). Comparative genomics boosts target predic-
tion for bacterial small RNAs. Proceedings of the Na-
tional Academy of Sciences, 110(37):E3487–96.
APPENDIX
A Benchmarking Workflow
The benchmark workflow used for the creation of the
prediction accuracy plots is shown in Figure 8. First,
for each sRNA query, optimal interactions with all
mRNA targets are predicted using INTARNA with a
certain parameter set. Then the resulting interactions
are sorted according to their energy values, from the
most favourable, i.e. those with the lowest values, to
the least favourable. Finally, we identify the rank of
each verified target (considered a supported predic-
tion). For each maximal rank value (later plotted on
the x-axis), we count the number of verified targets for
all sRNA queries that show a rank smaller or equal to
this.
This process is repeated for each benchmarked pa-
rameter set. The data collected is then plotted. The
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
138

x-axis of the plot represents the maximal rank we
consider for each result file, e.g. a number of target
predictions of 20 means that we consider the 20 first
lines of each result file and count how many verified
targets appear. This is covered by the y-axis, which
represents how many verified targets were among the
considered top-ranked target predictions. The violin
plots are generated by calculating the difference of a
certain parameterization of INTARNA e.g. c
B
= 11 to
the original predictor. This reveals general trends of
the different methods compared to the curve plot.
mRNA
targets
sRNA 1
IntaRNA
Result
file 1
sort
energy
sorted
file 1
mRNA
targets
sRNA 2
IntaRNA
Result
file 2
sort
energy
sorted
file 2
rank 7
rank 15
rank 202
verified
targets
Figure 8: Depiction of the benchmarking process.
Constraint Maximal Inter-molecular Helix Lengths within RNA-RNA Interaction Prediction Improves Bacterial sRNA Target Prediction
139

B Formal Recursions
E
S
(i, j, k, l) =
energy contribution for
stacking base pairs (i, j), (k, l)
!
: if k − i = 1 and l − j = 1,
+∞ : otherwise
(3)
E
IL
(i, j, k, l) =
energy contribution for
stack or interior loop (i, j), (k, l)
!
: if i < k and j < l,
+∞ : otherwise
(4)
helix(i, j,k,l) =
min
(
E
S
(i, j, i + 1, j + 1) + helix(i + 1, j + 1,k,l)
E
S
(i, j, k, l)
: if canonical helix
helix
U
(i, j, k, l;c
U
,c
B
) : otherwise
(5)
helix
U
(i, j, k, l;U,B) = min
min
p,q with
min(k−p,l−q)≥B−1
(p−i)+(q− j)≤U +2
E
IL
(i, j, p,q)
+helix
U
(p, q,k,l;U, B − 1)
!
: if B > 2,
E
IL
(i, j, k, l) : otherwise.
(6)
helix
S
(i, j, k, l) = min
p,r
q,s
helix(i, j, p,q) + seed(p,q,r,s) + helix(r,s,k,l)
(7)
H(i, j,k, l) = min
helix(i, j,k,l) + E
init
min
p,r,q,s with
(r−p)+(s−q)>c
U
+2
helix(i, j,k,l)≤E
helix
max
helix(i, j, p,q) + E
IL
(p, q,r,s) + H(r,s,k,l)
(8)
H
S
(i, j, k, l) = min
helix
S
(i, j, k, l) + E
init
min
p,r,q,s with
(r−p)+(s−q)>c
U
+2
helix
U
(i, j,k,l)≤E
helix
max
helix
S
(i, j, p,q) + E
IL
(p, q,r,s) + H(r,s,k,l)
min
p,r,q,s with
(r−p)+(s−q)>c
U
+2
helix(i, j,k,l)≤E
helix
max
helix(i, j, p,q) + E
IL
(p, q,r,s) + H
S
(r, s,k,l)
(9)
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
140
