
Functional Annotation of Proteins using Domain Embedding based
Sequence Classification
Bishnu Sarker
a
, David W. Ritchie and Sabeur Aridhi
University of Lorraine, Inria, Loria, CNRS, F-54000, Nancy, France
Keywords:
Machine Learning, Representation Learning, Protein Function Annotation, Bioinformatics, Domain
Embedding.
Abstract:
Due to the recent advancement in genomic sequencing technologies, the number of protein sequences in pub-
lic databases is growing exponentially. The UniProt Knowledgebase (UniProtKB) is currently the largest and
most comprehensive resource for protein sequence and annotation data. The May 2019 release of the Uniprot
Knowledge base (UniprotKB) contains around 158 million protein sequences. For the complete exploita-
tion of this huge knowledge base, protein sequences need to be annotated with functional properties such as
Enzyme Commission (EC) numbers and Gene Ontology terms. However, there is only about half a million
sequences (UniprotKB/SwissProt) are reviewed and functionally annotated by expert curators using informa-
tion extracted from the published literature and computational analyses. The manual annotation by experts are
expensive, slow and insufficient to fill the gap between the annotated and unannotated protein sequences. In
this paper, we present an automatic functional annotation technique using neural network based based word
embedding exploiting domain and family information of proteins. Domains are the most conserved regions
in protein sequences and constitute the building blocks of 3D protein structures. To do the experiment, we
used fastText
1
, a library for learning of word embeddings and text classification developed by Facebook’s AI
Research lab. The experimental results show that domain embeddings perform much better than k-mer based
word embeddings.
1 INTRODUCTION
1.1 Protein Function Annotation
Proteins are important components of all biological
systems. Understanding protein function is one of
the keys to understanding life at the molecular level,
and is central to understanding disease processes and
guiding drug discovery efforts (Bakheet and Doig,
2009). In this age of rapid and affordable amino-acid
sequencing technologies, the number of protein se-
quences accumulating in public databases is rising at
an increasing rate (Berger et al., 2016). In order to
enrich and exploit this huge quantity of data, these se-
quences should be annotated with functional proper-
ties. The UniProt knowledge base (UniProtKB) (The
UniProt Consortium, 2015) is currently the largest
public sequence database. It consists of two compo-
nents: (i) the UniProtKB/Swiss-Prot database which
contains protein sequences with reliable information
a
https://orcid.org/0000-0002-9716-4989
1
https://github.com/facebookresearch/fasttext
that has been reviewed by expert bio-curators, and (ii)
the UniProtKB/TrEMBL database that stores unanno-
tated sequences. Thus, for all proteins in UniProtKB,
we have the primary amino acid sequences as well
as some further information such as InterPro domain
definitions which may have been identified from fam-
ilies of similar sequences or 3D protein structures.
Enzymes are usually labelled following the En-
zyme Commission (EC) system (Cornish-Bowden,
2014), the widely used numerical enzyme classifica-
tion scheme. The EC System assigns each enzyme
a four digits number. The challenge, therefore, is to
annotate the un-annotated protein sequences automat-
ically with appropriate EC number. The EC classifi-
cation system has a hierarchical structure. The first
level consists of the six main enzyme classes: (i) ox-
idoreductases, (ii) transferases, (iii) hydrolases, (iv)
lyases, (v) isomerases and (vi) ligases, represented by
the first digit. Each main class node further extends
out several subclass nodes, specifying sub classes of
the enzymes, represented by the second digit. Simi-
larly, the third digit indicates the sub-subclass and the
fourth digit denotes the sub-sub-sub classes. Let us
Sarker, B., Ritchie, D. and Aridhi, S.
Functional Annotation of Proteins using Domain Embedding based Sequence Classification.
DOI: 10.5220/0008353401630170
In Proceedings of the 11th International Joint Conference on Knowledge Discovery, Knowledge Engineering and Knowledge Management (IC3K 2019), pages 163-170
ISBN: 978-989-758-382-7
Copyright
c
2019 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
163

consider as an example a Type II restriction enzyme,
which is annotated as EC 3.1.21.4. The first digit, 3,
denotes that it is a hydrolase. The second digit, 1, in-
dicates that it acts on ester bonds. The third digit, 21,
shows that it is an endodeoxyribonuclease producing
5-phosphomonoesters. The last digit, 4, specifies that
it is a Type II site-specific deoxyribonuclease.
1.2 Text Classification
One of the important tasks in natural language pro-
cessing is to classify text into classes such as tags, cat-
egories, labels, and so on. Text classification is widely
used in web search, information retrieval, ranking
and document classification. Due to recent successes,
neural network based models are prevalent in text
classifications. Although the representation capabil-
ity of neural networks is higher, training neural net-
work based deep learning models are computationally
expensive due to the presence of non-linear hidden
layer (Mikolov et al., 2013; Joulin et al., 2017). On
the other hand, linear classifiers are simple and effi-
cient, yet achieve better performance in the context
of text classification. However, linear classifier like
SVM does not share parameters among features and
classes (Joulin et al., 2017), which eventually limits
the generalization power of linear classifier. The fast-
Text(Joulin et al., 2016) text classification tool uses
log-linear model with a shallow neural network to
build a simple, fast, and efficient text classifier with
word embedding.
1.3 Text Classification for Protein
Function Annotation
Natural language text posses a defined linguistic
structure containing an array of words delimited by
various punctuation marks. Whereas biological texts
such as protein sequences are strings of letters se-
lected from an alphabet consists of 20 letters, each
representing an amino acid (Kimothi et al., 2016). Es-
sentially one string stands for a single protein. Un-
like natural texts, there is no way of formally defin-
ing words or phrases in protein sequences. Therefore,
using a text classification model requires further pre-
processing of protein sequences. The most common
way of pre-processing is to break the sequences into
biological words commonly known as K-mers that are
smaller units of size k composed of consecutive al-
phabets. The pre-processing can be done in two dif-
ferent ways: 1) Overlapping k-mers achieved by mov-
ing a k-size window over the sequence. For exam-
ple, let us break ”MAPPSV FSEV ” into overlapping
3-mers. The corresponding 3-mers are MAP, APP,
PPS, PSV, SV F, V FS, FSE, and SEV . Therefore,
the biological sequence ”MAPPSV FSEV ” is trans-
formed into following space delimited text: MAP
APP PPS PSV SVF VFS, FSE, SEV; (2) Using non-
overlapping k-mers, k numbers of sequences are gen-
erated by splitting the original sequences into non-
overlapping words of k consecutive letters with a
starting position moved by one letter for each newly
generated sequence (Asgari and Mofrad, 2015; Ki-
mothi et al., 2016). For example, for the sequence
”MAPPSV FSEV ”, considering 3-mers, the 3 newly
generated space delimited sequences are as follows:
1. MAP PSV FSE
2. APP SVF SEV
3. PPS VFS
Non-overlapping K-mers have been used for learn-
ing word embedding tasks and have been shown to
have better prediction accuracy when applied to fam-
ily classification task (Asgari and Mofrad, 2015).
Overlapping K-mers are widely used in homology
based sequence search in large databases of protein
sequences like (Altschul et al., 1997).
This paper presents a novel way to tokenize the
protein sequences for the purpose of functional anno-
tation. Instead of k-mers, we use domain and fam-
ily information of protein in order to learn protein
domain embeddings. The rationale behind using do-
main information is that the domains are the meaning-
ful units of protein sequence conserved across simi-
lar sequences. Domains may be considered as natural
building blocks of proteins. Due to evolution, pro-
tein domains may have gone through changes such
as duplication, fusion, recombination to produce pro-
teins with distinct structures and functions (Kummer-
feld and Teichmann, 2009). On the other hand, k-
mer words do not carry any biologically significant
meaning by themselves. Thus, tokenizing a protein
sequence into sentence of domains is more informa-
tive than sentence of k-mer words. The experimen-
tal results presented in experimental section verify the
higher performance of domain embeddings.
Two steps are necessary to prepare a training cor-
pus using domain information:
(1) Firstly, for each of the sequences, identify
domain signatures using InterProScan (Jones et al.,
2014; Quevillon et al., 2005; Mitchell et al., 2018),
which is a sequence analysis software that integrates
different protein signature recognition methods into
one resource. InterProScan provides domain signa-
tures along with their location of appearance in the
sequence.
(2) Secondly, InterProScan output is processed to
collect the domain signatures and sorting them ac-
KDIR 2019 - 11th International Conference on Knowledge Discovery and Information Retrieval
164

cording to their location of appearance. The domains
are organized in ascending order of their starting po-
sition in the sequence to form the domain-sentence.
Thus each line of the final corpus is a list of domains
found by InterProScan for a given protein sequence
along with true EC labels.
In this paper, we propose an automatic protein
function annotation technique that uses a shallow neu-
ral network based text classification method based on
domain embeddings. To accomplish the task, We
have used fastText developed by Facebook Artificial
Intelligence Research team to train a supervised se-
quence classification model as well as domain em-
beddings. We show a comparative analysis of over-
lapping 3-mers, non-overlapping k-mers and domain
embeddings for protein function annotation with en-
zyme commission number.
The remainder of the paper is organized as fol-
lows. Section 2 presents a brief summary of the previ-
ous works. Section 3 describes the proposed domain
embedding based method for automatic protein func-
tion annotation. Section 4 describes our experimental
setup and the obtained results.
2 PREVIOUS WORK
To annotate the protein sequences, the UniProt cu-
rators use two systems: (1) UniRule, uses a man-
ually designed if-then rules (Gattiker et al., 2003),
and (2) the Statistical Automatic Annotation System
(SAAS) (Kretschmann et al., 2001), which uses de-
cision tree based algorithm to automatically gener-
ate UniRule rules. Several approaches for predict-
ing Enzyme Commission (EC) numbers that exploit
structural and sequence similarities of proteins have
been described previously (Dobson and Doig, 2005;
Yang et al., 2015; Nagao Chioko and Kenji, 2014;
Rahman et al., 2014; Kumar and Skolnick, 2012;
Quester and Schomburg, 2011; Yu et al., 2009). Ad-
ditionally, machine learning methods have also been
used extensively in (des Jardins et al., 1997; Na-
gao Chioko and Kenji, 2014; Li et al., 2016; Huang
et al., 2007; Lu et al., 2007; Nasibov and Kandemir-
Cavas, 2009; Li et al., 2018; Shen and Chou, 2007).
For example, DEEPre (Li et al., 2018) predicts EC
numbers combining multiple tools and techniques in-
cluding PSI-Blast (Altschul et al., 1997), HMMER
(Finn et al., 2011), Convolutional and Recurrent Neu-
ral Networks, and sequence encoding using position
specific scoring matrix (PSSM) to perform dimen-
sionality uniformization, feature selection, and clas-
sification model training. EzyPred (Shen and Chou,
2007) predicts enzyme functional classes and sub-
classes using a top-down approach. EzyPred exploits
functional and evolutionary information of proteins
using pseudo amino acid composition (Chou, 2009)
and functional encoding. Based on two features,
EzyPred uses a modified K-Nearest Neighbor Classi-
fier called OET-KNN (Optimized Evidence-Theoretic
K-Nearest Neighbour).
SVM-Prot is a support vector machine (SVM)
based classification method detailed in (Cai et al.,
2003; Cai et al., 2004; Cai and Chou, 2005) and later
updated by adding two more classifiers, K-Nearest
Neighbor (KNN) and Probabilistic Neural Networks
(PNN) for improved performance in 2016 (Li et al.,
2016). This approach transforms protein sequences to
numerical representations based on various physico-
chemical properties such as polarity, hydrophobicity,
surface tension, charge, normalized van der Waals
volume, polarizability, secondary structure, solvent
accessibility, molecular weight, solubility, and the
numbers of hydrogen bond donors and acceptors in
side chain atoms.
ECPred (Dalkiran et al., 2018) is a hierarchical
prediction approach that starts by assigning the query
sequence as either an enzyme or non-enzyme. If
the query sequence is predicted to be an enzyme,
ECPred predictions follow the hierarchy of the EC
numbering system. Recently published high perform-
ing machine learning based approaches like DEEPre,
ECPred trains many models to accomplish the hier-
archical predictions. They train one model for each
class. For example, in DEEPre, one model to distin-
guish enzyme and non-enzyme, then if enzyme, an-
other model to predict the main class and 6 models
to predict the second digit of the EC and so on. The
works in (Kimothi et al., 2016; Asgari and Mofrad,
2015; Matsuda et al., 2005) presents unsupervised
word embedding based protein classification tech-
niques using continuous bag of words (CBOW) and
Skip-gram model proposed by (Mikolov et al., 2013).
(Asgari and Mofrad, 2015) explores non-overlapping
3-mer embeddings and applied the method for pro-
tein family classification. Although, they show an
improved performance for protein domain classifica-
tion, they have not explored the functional annotation
of proteins. In this article, we explore the protein do-
main embedding for functional classification of pro-
teins. We show that the proposed method performs
with higher accuracy than 3-mer embeddings in func-
tional annotation of protein using EC Number. Here
we learn single model to predict the complete EC an-
notation thus the proposed method does not incur the
overhead of learning many class-based hierarchical
models.
Functional Annotation of Proteins using Domain Embedding based Sequence Classification
165
.
.
.
1
0
0
0
.
.
.
0
0
0
1
.
.
.
0
1
0
0
.
.
.
.
.
1
0
0
.
.
.
0
0
0
h1
x1
x
c
x2
INPUT
W
VxN
W
VxN
W
VxN
N-Dimension
Hidden
Layer
OUTPUT
SOFTMAX
W
'
Nxv
Y
.
.
.
Figure 1: Continuous Bag of Words Model Architecture
(Mikolov et al., 2013).
3 METHODS
Our proposed enzyme classification technique is
based on supervised text classification model for nat-
ural language processing. The supervised classifica-
tion model is designed in a similar way to Continuous
Bag of Words (CBOW) described in (Mikolov et al.,
2013) architecture, where the middle word is replaced
by the label. The typical CBOW model architecture
is shown in Fig. 1. CBOW employs very simple neu-
ral network with single hidden layer for learning the
projection of individual word. In supervised mode,
for N textual documents or sentences, the following
negative log-likelihood is optimized over the classes
as described in (Joulin et al., 2017):
−
1
N
N
∑
n=1
y
n
log( f (W
0
W x
n
)),
Where W is a weight matrix that serves as a look-up
table over the words connecting input to the hidden
layer, W
0
is a weight matrix that connects hidden layer
with output layer where softmax function f is applied
to compute the probability distribution of the labels.
x
n
is the normalized bag of words of the n-th docu-
ment. y
n
is the label under consideration.
The discretely learned word representations are
averaged to learn the text representation which is then
fed into a linear classifier. The model uses stochastic
gradient descent based back propagation for optimiz-
ing the loss function.
To apply this model to enzyme classification, we
have used domain and family signatures of proteins
as discrete words. The individual domain signatures
found by running InterProscan against each protein
sequence serves the purpose of words. While prepar-
ing the training corpus, the domains are arranged in
ascending order of their location of appearance in the
sequence. For the comparison purposes, we have also
used overlapping and non-overlapping k-mer based
biological words processed from sequence data. The
Enzyme Commission (EC) numbers are used as labels
without any further processing.
After pre-processing the protein sequences to gen-
erate domains and K-mers and associating appropri-
ate EC labels, we prepared a large corpus of biological
texts for the purpose of learning embeddings and clas-
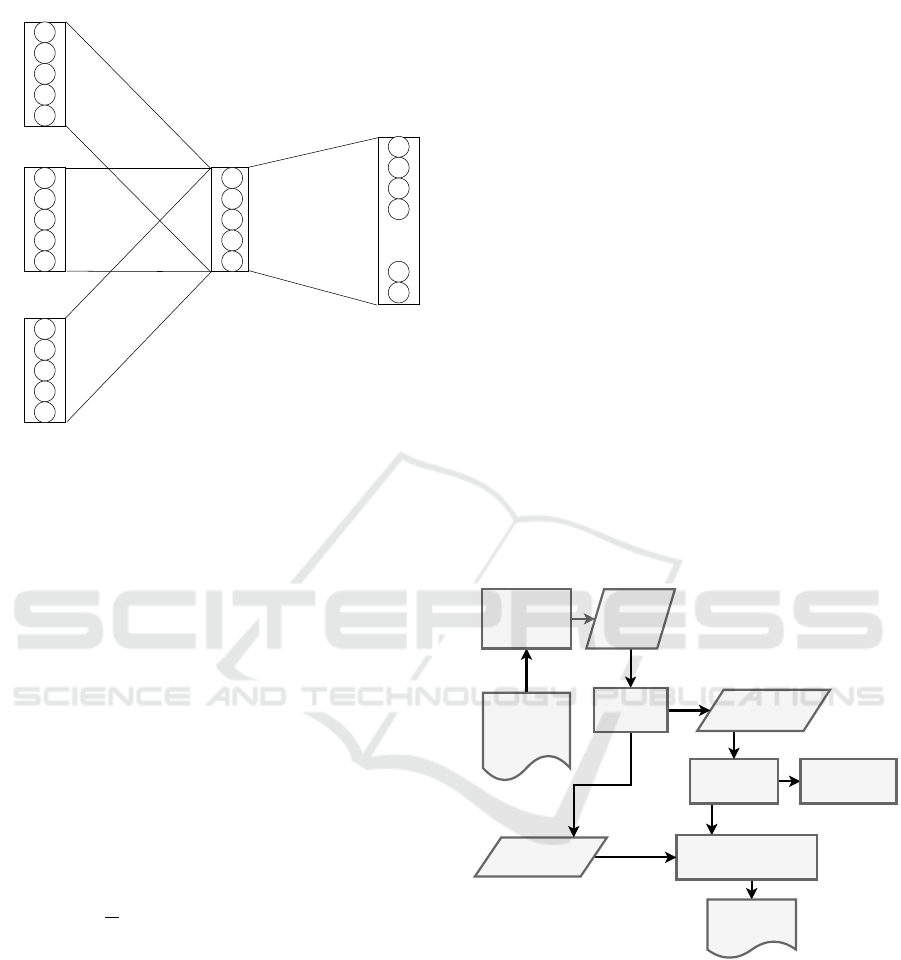
sification models. The work flow for the classification
task is shown in Fig:2. This figure shows the steps in-
volved in training a domain embedding model using
fastText supervised learning. The raw sequence data
is transformed into domain data using InterProScan
(Jones et al., 2014) sequence analysis tool and then
feed into a supervised learning model to learn the em-
beddings and classification model. Finally, the test
data are feed into the model to predict the EC annota-
tions.
Fasttext
Supervised
Learning
Domain
Embeddings
Classification Model
using Domain
Embeddings
Predicted
Annotations
Protein
Sequences
and
Labels
Pre-processing
using
InterProScan
Protein
Domain
Corpus
Training Set
Test Set
Splitting
Corpus
Figure 2: Data preparation and training work-flow for Do-
main Embedding based Protein Function Annotation.
4 EXPERIMENTS AND RESULT
ANALYSIS
In this Section, we first present the used data. Then,
we present our experimental protocol and we discuss
the obtained results.
KDIR 2019 - 11th International Conference on Knowledge Discovery and Information Retrieval
166

4.1 Dataset and Training
To evaluate the method, we have used a well defined
dataset of enzyme and non-enzyme proteins curated
from UniprotKB (The UniProt Consortium, 2015).
This dataset is called “NEW” and was published by
(Li et al., 2018) and was constructed as follows:
1. The SWISS-PROT (released on September 7,
2016) database was separated into enzymes and
non-enzymes based on their annotation.
2. To guarantee uniqueness and correctness, enzyme
sequences with more than one set of EC num-
bers or incomplete EC number annotation were
excluded.
3. To avoid fragment data, enzyme sequences anno-
tated with ‘fragment’ or with less than 50 amino
acids were excluded. Enzyme sequences with
more than 5000 amino acids were also excluded.
4. Redundancy bias is removed using CD-HIT (Fu
et al., 2012) with 40% similarity threshold to sift
the raw dataset, resulting in 22,168 low-homology
enzyme sequences.
5. To construct the non-enzyme part, 22,168 non-
enzyme protein sequences were randomly col-
lected from the SWISS-PROT (released on
September 7, 2016) non-enzyme part, which were
also subject to the above (ii–iv) steps. Thus the
original dataset contains 22,168 enzymes and an
equal number of non-enzymes.
To build the corpus for learning the embeddings,
each sequence has undergone three different pre-
processing treatment for 3 different types of sequence
tokenization described in the section 1.3. For over-
lapping k-mer, each sequence is split into overlapping
K-mer words. We have chosen 3-mer for our experi-
ment meaning a window of size 3 is moved over the
sequence to split it into overlapping 3-mers.
For non-overlapping k-mer, each sequence was
transformed into 3 sequences of non-overlapping 3-
mers. And same label is associated with all of the
3 sequences as they are generated from the same se-
quence with single EC label.
In the last case, we used domain and family
signatures of proteins as words to build the train-
ing corpus. Each sequence was transformed into
a list of domains using InterProScan(Jones et al.,
2014) software of Version 5.35-74.0. We built 3
large corpus using the 3-mers (overlapping and non-
overlapping) and domains and each sentence is asso-
ciated with EC labels to train the supervised classi-
fication model. For non-enzyme, we have labelled
them with ”
label NANZ”.
For the implementation purpose, we used fast-
Text(Joulin et al., 2016) which is library for learn-
ing of embeddings and text classification developed
by facebook’s AI research.
4.2 Evaluation
To evaluate our method, we used 10-fold cross-
validation on NEW Dataset. We split the text cor-
pus into 10 parts, and considered one of them as a
testing set with the remaining 9 parts composedly be-
ing used as the training set. The cross validation re-
sults show promising performance of the domain em-
bedding based Enzyme classification. For each query
sequence, we picked the top ranked annotation only.
To validate the performance, we computed the accu-
racy, macro-precision, macro-recall, and macro-F1-
measure at different levels of EC number. The fol-
lowing formulae (as used in (Li et al., 2018)) were
used to compute the evaluation metrics:
accuraccy(y, y
0
) =
1
N
N−1
∑
i=0
1(y
i
= y
0
i
),
Here, y and y
0
are the list of ground truths and pre-
dicted annotations. The accuracy is computed for
each level of EC annotation. As the problem is a mul-
ticlass classification problem, we computed macro-
precision, macro-recall, and macro-F1 score as fol-
lows:
Macro − precision(y, y
0
) =
1
|M|
∑
l∈M
precision(y
l
, y
0
l
),
Macro − recall(y, y
0
) =
1
|M|
∑
l∈M
recall(y
l
, y
0
l
),
Macro − F1(y, y
0
) =
1
|M|
∑
l∈M
F1 measure(y
l
, y
0
l
),
Here, y
l
is the part of y with the label l and y
0
l
is the
part of y
0
with label l. And M is the set of classes.
In general the precision, recall, and F1-Measure are
computed as follows when two sets A and P are given:
precision =
|A ∩ P|
|P|
,
recall =
|A ∪ P|
|A|
,
F1 − measure =
2 × precision × recall
precision + recall
.
Here, A is the set of ground truths and P is the set of
predictions. As EC numbers are hierarchical with 4
levels, we report level-wise precision, recall and F1-
measure. Level-1 denotes main class, level-2 denotes
Functional Annotation of Proteins using Domain Embedding based Sequence Classification
167

Table 1: The experimental results of 10-fold Cross-Validation on ”NEW” dataset.
LEVEL EMBEDDINGS ACCURACY MACRO-PRECISION MACRO-RECALL MACRO-F1
LEVEL-0
3-MER NON-OVERLAP 0.741 0.768 0.776 0.772
3-MER OVERLAP 0.738 0.816 0.825 0.82
DOMAIN 0.943 0.968 0.97 0.969
LEVEL-1
3-MER NON-OVERLAP 0.523 0.243 0.24 0.241
3-MER OVERLAP 0.529 0.251 0.245 0.248
DOMAIN 0.929 0.918 0.913 0.916
LEVEL-2
3-MER NON-OVERLAP 0.467 0.09 0.089 0.089
3-MER OVERLAP 0.47 0.101 0.097 0.099
DOMAIN 0.918 0.823 0.812 0.817
LEVEL-3
3-MER NON-OVERLAP 0.452 0.062 0.06 0.061
3-MER OVERLAP 0.455 0.073 0.07 0.071
DOMAIN 0.909 0.757 0.757 0.757
LEVEL-4
3-MER NON-OVERLAP 0.434 0.037 0.036 0.037
3-MER OVERLAP 0.439 0.052 0.05 0.051
DOMAIN 0.851 0.62 0.633 0.626
sub-class, level-3 denotes sub-sub-class and level-4
denotes sub-sub-sub class. We add level-0 to the step
of identifying enzyme or non-enzyme. For each query
sequence, we pick the top-most annotation. For eval-
uation purposes, we split the 4-digit EC annotation
into its constituent parts. Then, for level-1 we con-
sider first digit, for level-2 we take first 2 digits, for
level-3 we take first 3-digits and finally for level-4 we
take all four digits together.
In our experiments, we have trained the model us-
ing 0.9 as learning rate, 150 hidden units, window size
of 5 and trained for 200 epochs. We have used Hier-
archical Softmax as loss function and word n-grams
of 2. The cross validation result is shown in the table
1.
Table-1 shows the accuracy, precision, recall and
F1 measure of the cross-validation performed over the
NEW dataset. As EC numbers consist of four dig-
its, we present the evaluation metrics for four lev-
els (level-1, 2, 3, 4). To measure the efficiency
in classifying enzymes and non-enzymes among the
test sequences, we present level-0 accuracy, preci-
sion, recall and F1 measure. The result shows that
the proposed domain embedding based classification
can differentiate enzymes and non-enzymes with an
accuracy of 94.3% which is better than k-mer based
embeddings(73%-74%). Along with accuracy, we
also report the macro precision, recall, and F1 score
weighted over classes. Macro-{precision, recall and
F-1} scores give a reliable measure for unbalanced
data. Domain embeddings perform with very high
precision and recall of 97% for level-0 prediction
task. It is interesting to see that the macro-F1 score
confirms the higher accuracy shown by the proposed
domain embeddings.
For level-1 predictions, we predict the main class
if the protein is an enzyme. There are 6 different main
classes. We measure the level-1 accuracy as how ac-
curately it can identify the non-enzymes and enzymes
with the correct main class. Similar to level-0, we also
present the class based macro-{precision, recall, and
F1} scores as the test data is class imbalanced. The
performance measures show that domain embedding
based classification performs better in all metrics.
In a similar fashion, we also report the accuracy,
macro-{precision, recall and F1} scores for level-2,
level-3 and level-4 predictions. For all of the levels,
our proposed annotation technique outperforms K-
mer based embeddings. However, as we go to higher
EC levels, the accuracy falls off because a higher EC
level is very specific. For example, a Level-4 EC
number describes an enzyme that is specific for a par-
ticular type of substrate molecule. However, the do-
main embeddings based classification shows promis-
ing performance for level-4 prediction also.
In summary, from the results shown in Table 1,
it is evident that domain based embeddings perform
noticeably better than k-mer based word embeddings
in all evaluation metrics and also for all levels of EC
hierarchy.
5 CONCLUSION
In this paper, we propose a novel protein function an-
notation approach using domain embedding based se-
quence classification instead of k-mer based word em-
bedding. To show the superior performance of the
proposed method, we used 10-fold cross-validation
on benchmark dataset. We measured the annotation
KDIR 2019 - 11th International Conference on Knowledge Discovery and Information Retrieval
168

performance using accuracy and we also report the
macro precision, macro-recall, and macro-F1 mea-
sure to reduce the effect of class imbalance in the
test dataset. According to all the evaluation metrics
we considered, the proposed approach show better
performance. One of the strengths of the proposed
method lies in it’s simplicity. The method learns do-
main embeddings using a single layer neural network.
Due to the use of shallow neural network, the training
is faster than other multi-layer deep networks. We
have used hierarchical softmax loss function to make
training even faster. Unlike other hierarchical clas-
sification models like ECPred(Dalkiran et al., 2018)
and DEEPre(Li et al., 2018), the proposed method
learns single model instead of learning many models
each for every class. The method is scalable for larger
dataset using CUDA based GPU units. Although the
proposed method performs well, there is still scope of
improvement specially for level-3 and level-4 predic-
tions. As a future plan, we envision to improve the
method for more precise predictions and also to apply
the similar approach for protein function annotation
using Gene Ontology Terms.
REFERENCES
Altschul, S. F., Madden, T. L., Sch
¨
affer, A. A., Zhang,
J., Zhang, Z., Miller, W., and Lipman, D. J. (1997).
Gapped blast and psi-blast: a new generation of pro-
tein database search programs. Nucleic Acids Re-
search, 25(17):3389–3402.
Asgari, E. and Mofrad, M. R. (2015). Continuous
distributed representation of biological sequences
for deep proteomics and genomics. PloS one,
10(11):e0141287.
Bakheet, T. M. and Doig, A. J. (2009). Properties and iden-
tification of human protein drug targets. Bioinformat-
ics, 25(4):451–457.
Berger, B., Daniels, N. M., and Yu, Y. W. (2016). Com-
putational biology in the 21st century: Scaling with
compressive algorithms. Commun. ACM, 59(8):72–
80.
Cai, C., Han, L., Ji, Z., and Chen, Y. (2004). Enzyme family
classification by support vector machines. Proteins:
Structure, Function, and Bioinformatics, 55(1):66–76.
Cai, C., Han, L., Ji, Z. L., Chen, X., and Chen, Y. Z.
(2003). Svm-prot: web-based support vector ma-
chine software for functional classification of a protein
from its primary sequence. Nucleic acids research,
31(13):3692–3697.
Cai, Y.-D. and Chou, K.-C. (2005). Predicting enzyme sub-
class by functional domain composition and pseudo
amino acid composition. Journal of Proteome Re-
search, 4(3):967–971.
Chou, K.-C. (2009). Pseudo amino acid composition and its
applications in bioinformatics, proteomics and system
biology. Current Proteomics, 6(4):262–274.
Cornish-Bowden, A. (2014). Current IUBMB recommen-
dations on enzyme nomenclature and kinetics. Per-
spectives in Science, 1(1-6):74–87.
Dalkiran, A., Rifaioglu, A. S., Martin, M. J., Cetin-Atalay,
R., Atalay, V., and Do
˘
gan, T. (2018). ECPred: a tool
for the prediction of the enzymatic functions of pro-
tein sequences based on the EC nomenclature. BMC
Bioinformatics, 19(1):334.
des Jardins, M., Karp, P. D., Krummenacker, M., Lee, T. J.,
and Ouzounis, C. A. (1997). Prediction of enzyme
classification from protein sequence without the use
of sequence similarity. In Proc Int Conf Intell Syst
Mol Biol, volume 5, pages 92–99.
Dobson, P. D. and Doig, A. J. (2005). Predicting enzyme
class from protein structure without alignments. Jour-
nal of molecular biology, 345(1):187–199.
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER
web server: interactive sequence similarity searching.
Nucleic Acids Research, 39(2):W29–W37.
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). Cd-
hit: accelerated for clustering the next-generation se-
quencing data. Bioinformatics, 28(23):3150–3152.
Gattiker, A., Michoud, K., Rivoire, C., Auchincloss, A. H.,
Coudert, E., Lima, T., Kersey, P., Pagni, M., Sigrist,
C. J., Lachaize, C., Veuthey, A.-L., Gasteiger, E., and
Bairoch, A. (2003). Automated annotation of micro-
bial proteomes in SWISS-PROT. Computational Bi-
ology and Chemistry, 27(1):49–58.
Huang, W.-L., Chen, H.-M., Hwang, S.-F., and Ho, S.-
Y. (2007). Accurate prediction of enzyme subfam-
ily class using an adaptive fuzzy k-nearest neighbor
method. Biosystems, 90(2):405–413.
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W.,
McAnulla, C., McWilliam, H., Maslen, J., Mitchell,
A., Nuka, G., et al. (2014). Interproscan 5: genome-
scale protein function classification. Bioinformatics,
30(9):1236–1240.
Joulin, A., Grave, E., Bojanowski, P., Douze, M., J
´
egou,
H., and Mikolov, T. (2016). Fasttext.zip: Com-
pressing text classification models. arXiv preprint
arXiv:1612.03651.
Joulin, A., Grave, E., Bojanowski, P., and Mikolov, T.
(2017). Bag of tricks for efficient text classification.
In Proceedings of the 15th Conference of the Euro-
pean Chapter of the Association for Computational
Linguistics: Volume 2, Short Papers, pages 427–431.
Association for Computational Linguistics.
Kimothi, D., Soni, A., Biyani, P., and Hogan, J. M. (2016).
Distributed representations for biological sequence
analysis. arXiv preprint arXiv:1608.05949.
Kretschmann, E., Fleischmann, W., and Apweiler, R.
(2001). Automatic rule generation for protein anno-
tation with the C4.5 data mining algorithm applied on
SWISS-PROT. Bioinformatics, 17 10:920–6.
Kumar, N. and Skolnick, J. (2012). Eficaz2. 5: application
of a high-precision enzyme function predictor to 396
proteomes. Bioinformatics, 28(20):2687–2688.
Functional Annotation of Proteins using Domain Embedding based Sequence Classification
169

Kummerfeld, S. K. and Teichmann, S. A. (2009). Protein
domain organisation: adding order. BMC Bioinfor-
matics, 10(1):39.
Li, Y., Wang, S., Umarov, R., Xie, B., Fan, M., Li, L., and
Gao, X. (2018). DEEPre: sequence-based enzyme EC
number prediction by deep learning. Bioinformatics,
34(5):760–769.
Li, Y. H., Xu, J. Y., Tao, L., Li, X. F., Li, S., Zeng,
X., Chen, S. Y., Zhang, P., Qin, C., Zhang, C.,
et al. (2016). Svm-prot 2016: a web-server for ma-
chine learning prediction of protein functional fami-
lies from sequence irrespective of similarity. PloS one,
11(8):e0155290.
Lu, L., Qian, Z., Cai, Y.-D., and Li, Y. (2007). ECS: an au-
tomatic enzyme classifier based on functional domain
composition. Computational Biology and Chemistry,
31(3):226–232.
Matsuda, S., Vert, J.-P., Saigo, H., Ueda, N., Toh, H.,
and Akutsu, T. (2005). A novel representation of
protein sequences for prediction of subcellular loca-
tion using support vector machines. Protein Science,
14(11):2804–2813.
Mikolov, T., Sutskever, I., Chen, K., Corrado, G. S., and
Dean, J. (2013). Distributed representations of words
and phrases and their compositionality. In Advances in
neural information processing systems, pages 3111–
3119.
Mitchell, A. L., Attwood, T. K., Babbitt, P. C., Blum, M.,
Bork, P., Bridge, A., Brown, S. D., Chang, H.-Y., El-
Gebali, S., Fraser, M. I., et al. (2018). Interpro in
2019: improving coverage, classification and access
to protein sequence annotations. Nucleic acids re-
search, 47(D1):D351–D360.
Nagao Chioko, N. N. and Kenji, M. (2014). Prediction of
detailed enzyme functions and identification of speci-
ficity determining residues by random forests. PLoS
One, 9(1).
Nasibov, E. and Kandemir-Cavas, C. (2009). Efficiency
analysis of KNN and minimum distance-based clas-
sifiers in enzyme family prediction. Computational
Biology and Chemistry, 33(6):461–464.
Quester, S. and Schomburg, D. (2011). EnzymeDetec-
tor: an integrated enzyme function prediction tool and
database. BMC Bioinformatics, 12(1):376.
Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder,
N., Apweiler, R., and Lopez, R. (2005). InterProScan:
protein domains identifier. Nucleic Acids Research,
33(suppl
2):W116–W120.
Rahman, S. A., Cuesta, S. M., Furnham, N., Holliday, G. L.,
and Thornton, J. M. (2014). EC-BLAST: a tool to
automatically search and compare enzyme reactions.
Nature Methods, 11(2):171.
Shen, H.-B. and Chou, K.-C. (2007). Ezypred: a top–down
approach for predicting enzyme functional classes and
subclasses. Biochemical and biophysical research
communications, 364(1):53–59.
The UniProt Consortium (2015). UniProt: a hub for pro-
tein information. Nucleic Acids Research, 43(D204-
D212).
Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., and Zhang,
Y. (2015). The i-tasser suite: protein structure and
function prediction. Nature methods, 12(1):7.
Yu, C., Zavaljevski, N., Desai, V., and Reifman, J. (2009).
Genome-wide enzyme annotation with precision con-
trol: Catalytic families (CatFam) databases. Proteins:
Structure, Function, and Bioinformatics, 74(2):449–
460.
KDIR 2019 - 11th International Conference on Knowledge Discovery and Information Retrieval
170
