
BROGUE: A Platform for Constructing and Visualizing
“Gene-Mutation-Disease” Relation Knowledge Graphs to Support
Biomedical Research and Clinical Decisions
Dongsheng Zhao
1a
, Fan Tong
1b
, Zheheng Luo
2c
, Sheng Liu
3d
and Wei Song
3e
1
Information Center, Academy of Military Medical Sciences, Beijing, China
2
Information Department, No. 920 Hospital of PLA, Yunnan, China
3
Beijing MedPeer Information Technology Co., Ltd., Beijing, China
Keywords: Gene, Mutation, Disease, Knowledge Graph, Platform.
Abstract: In the era of precision medicine, clinicians need intensive and comprehensive evidence to conduct research
and make decisions. However, current knowledge bases are isolated and lack integration with information
from other databases or literature, constituting an obstacle for clinicians to locate and understand their
interested relations. In this paper, we design a platform development methodology to construct and visualize
a biomedical knowledge graph combining text mining tools and knowledge fusion models with web interface
libraries. The platform thereby provides the functions of knowledge acquisition, integration, storage, search
and visualization, where each concept in the relation is described by its properties, each relation in the database
is located to sentences and each paragraph in the article is translated into Chinese. To further validate the
feasibility and practicability, we applied the methodology to the “gene-mutation-disease” field and built a
Biomedical Relation of Gene-mUtation-diseasE (BROGUE) platform. The platform included 590 high-
quality gene-mutation-disease relations covering a wide range of commonly-used gene (286), mutation (525)
and disease (347) concepts by October 2019. Two tests demonstrated that BROGUE has potential to be useful
for supporting biomedical research and clinical decisions. The platform has been deployed and is publicly
available at http://brogue.medmdt.net/.
1 INTRODUCTION
Over more than two decades, evidence-based
medicine has rightfully become part of the fabric of
modern clinical practice and has contributed to many
advances in healthcare (McCartney et al., 2016),
eventually developing into several reliable systems,
including ClinicalKey (Huslig and Vardell, 2015),
UpToDate (Fox and Moawad, 2003) and medicine
(Meyers, 2000). However, the coming era of
precision medicine poses a challenge to "one size fits
all" evidence-based medicine owing to the latter’s
failure to provide adequate solutions for outliers
(Beckmann and Lew, 2016). Providing more
intensive and comprehensive evidence to the clinician
a
https://orcid.org/0000-0003-2616-8891
b
https://orcid.org/0000-0001-6636-8578
c
https://orcid.org/0000-0003-4516-5901
d
https://orcid.org/0000-0002-1054-6440
e
https://orcid.org/0000-0002-4596-5303
is critical for conducting scientific biomedical
research and making a timely clinical decision.
According to the National Research Council
(U.S.), precision medicine is defined as precisely
tailoring therapies to subcategories of disease on the
basis of genomics including genetic, biomarker,
phenotypic, or psychosocial characteristics (Ashley,
2015; Jameson and Longo, 2015). Hence, “gene-
mutation-disease” relations, as an essential
component of precision medicine, have been the
objects of significant study in recent years and several
high-quality biomedical knowledge bases such as
ClinVar (Landrum et al., 2013), COSMIC (Forbes et
al., 2014) and HGMD (Stenson et al., 2017) have
been built. These knowledge bases provide a large
amount of valuable data that have come to play
52
Zhao, D., Tong, F., Luo, Z., Liu, S. and Song, W.
BROGUE: A Platform for Constructing and Visualizing “Gene-Mutation-Disease” Relation Knowledge Graphs to Support Biomedical Research and Clinical Decisions.
DOI: 10.5220/0009154600520059
In Proceedings of the 13th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2020) - Volume 3: BIOINFORMATICS, pages 52-59
ISBN: 978-989-758-398-8; ISSN: 2184-4305
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
critical roles in supporting scientific research and
clinical decision-making.
There are nevertheless three major problems with
the above-mentioned existing knowledge bases:
construction efficiency, integration depth, and
presentation forms. First, to ensure the reliability and
validity of the knowledge, their creators have built
these knowledge bases using dedicated and
meticulous curation from domain experts with
medical training backgrounds and annotation
experience. The construction process is a highly
expensive and time-consuming endeavour, and it
becomes increasingly difficult as biomedical data and
findings rapidly grow. Second, different independent
institutions or organizations introduce each
knowledge base, so that these actors develop
knowledge bases under their own standards and
regulations during the process of construction and
maintenance. The lack of a usable and useful mapping
model creates difficulty bridging the gap between
these knowledge bases and relevant databases (e.g.
terminology and literature), further limiting the
coverage and depth of the knowledge. Third, current
knowledge bases display knowledge items using
tabulation, which involves clearly and directly
enumerating the involved biomedical concepts and
corresponding relation types. While this seems
advantageous from the perspective of relations
themselves, this item-level visualization fails to
present the overall picture of a relation network (i.e.
the connections and interactions between biomedical
concepts), at the same time omitting detailed
descriptions of each relation (e.g. subordinate
properties and supporting evidence).
We therefore designed a platform development
methodology for biomedical knowledge graph
construction as well as visualization, and developed a
platform named BROGUE (Biomedical Relation of
Gene-mUtation-diseasE) for feasibility and
practicability validation. Combining several cutting-
edge techniques and widely used tools, we integrated
relevant information about “gene-mutation-disease”
relations from diverse data sources. We provided this
comprehensive knowledge so as to help users locate
and understand their interested entities or relations.
The platform we present in this paper shows great
potential when it comes to inclusive knowledge
integration and sustainable knowledge discovery to
support biomedical research and clinical decisions.
2 METHODS
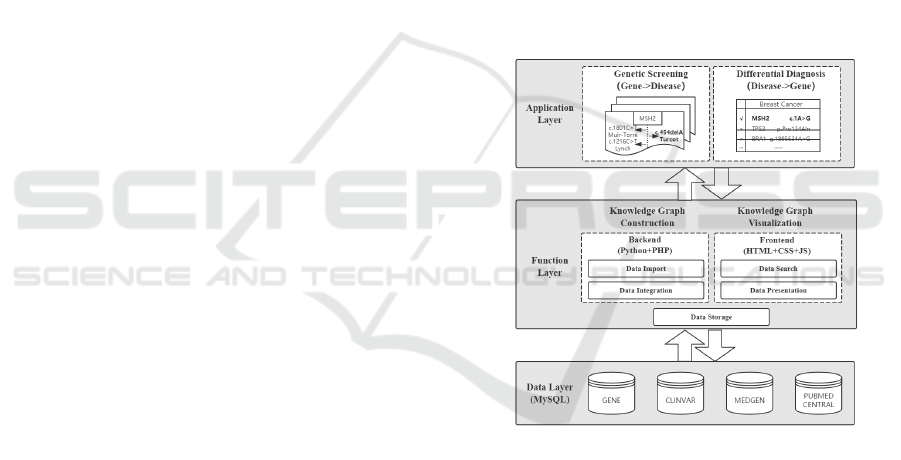
Figure 1 displays the overall architecture of our
platform. We built this platform on the WAMP Server
with the frontend implemented using HTML, CSS,
JavaScript, and the backend using PHP/Python and
the MySQL database. Functions currently
implemented by our platform include : 1) knowledge
acquisition which obtains relevant information from
distributed and heterogeneous data sources; 2)
knowledge integration which standardizes diverse
data into an intensive and comprehensive knowledge
graph; 3) knowledge storage which organizes linked
information into a structural and relational MySQL
database; 4) knowledge search which retrieves
relations and relation-associated metadata from a
constructed and integrated knowledge graph; and
finally 5) knowledge visualization which presents
relations, relation-related entities and relation-linked
articles with statistics and network graphs.
Figure 1: The overall architecture of BROGUE for
constructing and visualizing “gene-mutation-disease”
relation knowledge graph.
2.1 Knowledge Acquisition
We selected different authoritative databases from
NCBI as data sources of entity, relations and
literature, as Table 1 indicates. Among these
databases, ClinVar provides essential “gene-
mutation-disease” relation items to our platform,
while we introduced Gene (Maglott et al., 2005),
MedGen (Halavi et al., 2018) and PubMed Central
(Roberts, 2001) to assure and improve data integrity
and provenance tracking when it comes to those
relations. After downloading raw data from the NCBI
FTP server, we designed the following rules to filter
BROGUE: A Platform for Constructing and Visualizing “Gene-Mutation-Disease” Relation Knowledge Graphs to Support Biomedical
Research and Clinical Decisions
53

valid from less valuable data: 1) genes belong to
homo sapiens; 2) mutations cover substitution,
insertion, deletion, and InDel; 3) diseases consist of
abnormality, dysfunction, and syndrome; 4) relations
contain explicit gene, mutation, disease concepts with
at least one literature citation; 5) the ClinVar linking
literatures includes at least two types among gene,
mutation, and disease.
Table 1: Details for different types of data in BROGUE.
Data Type Data Source
Data
Scale
Data
URL
Gene Gene 60,932 1
Mutation ClinVa
r
222,786 2
Disease MedGen 50,578 3
Relation ClinVa
r
24,832 2
Literature PubMed Central 24,297 4
1.ftp://ftp.ncbi.nih.gov/gene/DATA/GENE_INFO/Mamm
alia/Homo_sapiens.gene_info.gz
2.ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/xml/archive/2018/
ClinVarFullRelease_2018-11.xml.gz
3.ftp://ftp.ncbi.nlm.nih.gov/pub/medgen/NAMES.RRF.gz
4.https://www.ncbi.nlm.nih.gov/pmc/
2.2 Knowledge Integration
We selected different authoritative databases centered
on “gene-mutation-disease” relations, then appended
entity properties (mutation type, allele source,
molecular consequence, gene type, and inheritance
type) and literature evidence to extend relation items.
On the one hand, facing inconsistency in entity
nomenclature, we built glossaries including standard
and synonymous mentions for gene, mutation and
disease entities (see Supplementary Information). We
mapped the entities involving the relations to the
concepts in corresponding terminology databases,
thus describing them by the properties of each
concept. On the other hand, instead of being satisfied
with article-level evidence, we constructed a relation
annotation pipeline combining automatic extraction
with expert curation. Under the supervision of the
ClinVar knowledge base, we located and labeled the
candidate entity co-occurrence sentences using text
mining tools, including NLTK (sentence
tokenization) (Loper and Bird, 2002), ezTag (named
entity recognition) (Kwon et al. 2018) and OpenIE
(relation extraction) (Saha, 2018) as well as a relation
mapping model introduced in previous work (Zhao,
Tong and Luo, 2019). Two independent domain
experts with medical training backgrounds
subsequently curated the annotation results, which
another expert would check further in a case of
inconsistent or cyclic judgment. Finally, the curated
results provide detailed sentence-level evidence and
rich context linked to “gene-mutation-disease”
relations.
2.3 Knowledge Storage
Instead of leveraging a graph database storage
solution (Vicknair et al., 2010), we proceeded with a
traditional relational database (MySQL, in this case)
similar to those used by ClinVar, COSMIC, and
HGMD. This is because MySQL meets the demand
of storage capability and query efficiency based on
the entity diversity and relation complexity of our
current “gene-mutation-disease” relation dataset. To
organize dispersed data from distributed and
heterogeneous data sources, we created association
tables for both entities (from terminology databases
to the ClinVar knowledge base) and relations (from
the literature database to the ClinVar knowledge
base) in addition to basic tables, such as dictionary
tables. Once having imported, cleaned and integrated
data from the previous process, our database finally
stores and indexes “gene-mutation-disease” relations
and related entities and articles information.
2.4 Knowledge Search
Once a query containing single entity or multiple
entities is submitted, the platform automatically
transforms non-standard expressions into standard
terminology using a bio-entities thesaurus built in the
previous step, then returns relations involving the
normalized form of searched entities. Unlike
independent gene and mutation concepts, the
intertwined disease notions are formed into a tree
structure. Our platform further extends the original
input disease terms to more specific and detailed
disease names beneath those terms according to the
hyponym hierarchy of MeSH vocabulary.
Simultaneously, the platform records user action to
calculate the searching frequency of each entity and
subsequently presents real-time trending on user
interests and preferences.
2.5 Knowledge Presentation
Not only are simply listed in tables according to the
user’s query, but the database also presents the
retrieved “gene-mutation-disease” relations in
various forms, including comprehensive knowledge
graphs and detailed literature annotations. For the
knowledge graphs, we took advantage of the D3
JavaScript library (King, 2014) to display overall
relation network where entities are treated as nodes
and relations are regarded as edges. In this context,
BIOINFORMATICS 2020 - 11th International Conference on Bioinformatics Models, Methods and Algorithms
54
the entities of gene, mutation and disease are
designated different colors and described with
different properties. Relation types are labelled on the
edge, which is linked to related literature. For
literature annotation, we designed customized CSS
styles to highlight the sentence-level location of the
relations scattered in the articles. In addition to
labelling the existing relations from the ClinVar
knowledge base, the platform marks novel relations
detected and discovered during the process of
extraction and curation, providing potentially
valuable information for further study. Moreover, we
introduced Google's neural machine translation
system (Wu et al., 2016) to help non-native English
users grasp a better understanding of the context
around the located relation. By implementing
sentences re-tokenization and terminology
substitution, we optimize the English-Chinese
translation results in the biomedical field.
3 RESULTS
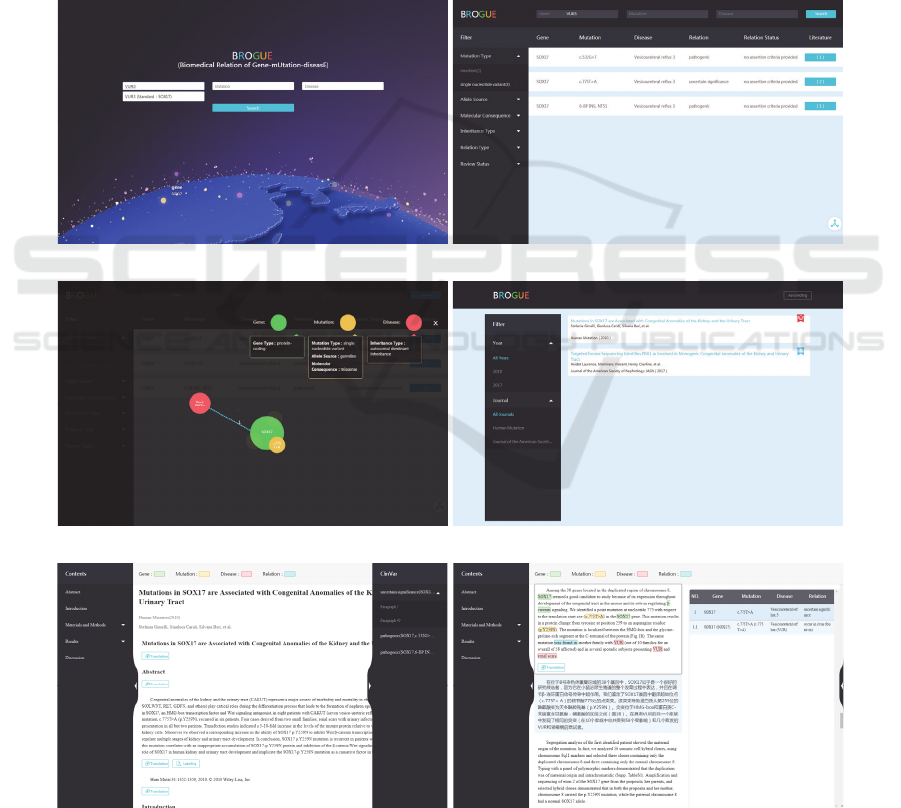
3.1 System Description
As Figure 2 illustrates, the graphical interface of our
web-based platform comprises 3 different kinds of
pages, including: 1) Search Page (Figure 2(a)) to
submit query of “gene-mutation-disease” relation;
(a) Relation search page (b) Relation search result page (table)
(c) Relation search result page (graph) (d) Linked literature result page
(e) Linked literature location page (f) Linked literature location page
Figure 2: The graphical user interface of BROGUE “gene-mutation-disease” relation knowledge graph.
BROGUE: A Platform for Constructing and Visualizing “Gene-Mutation-Disease” Relation Knowledge Graphs to Support Biomedical
Research and Clinical Decisions
55
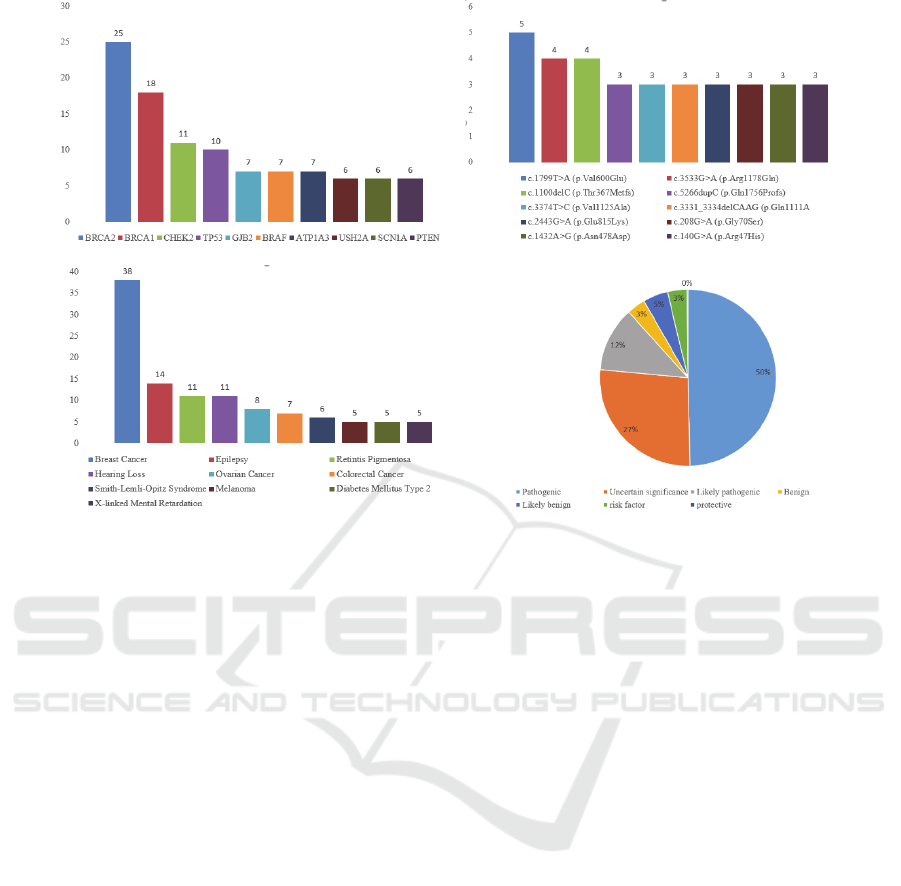
(a) Top 10 gene concepts of high frequency (b) Top 10 mutation concepts of high frequency
(c) Top 10 disease concepts of high frequency (d) Relation type distribution
Figure 3: The statistics of entities and relation covered in BROGUE.
2) Relation Presentation Page (Figure 2(b-c)) as the
terminal to enumerate and depict status and property
of “gene-mutation-disease” relation; 3) Literature
Visualization Page (Figure 2(d-f)) as the interface to
provide the location and description of “gene-
mutation-disease” relation. On our platform, users
can easily search and learn about interested entities or
relations through step-by-step operations. These
retrieval results may provide valuable and reliable
information that supports clinical research and
decision-making.
As Figure 3 demonstrates, our platform currently
includes 590 high-quality “gene-mutation-disease”
relations from ClinVar, covering commonly-used
gene (286), mutation (525) and disease (347)
concepts. The platform further linked relations to 527
full-length articles and located in 654 sentences
among these literatures. Beyond that, 25,375 entities
in the relation were described by their properties and
26,594 paragraphs in the article were translated into
Chinese. These were developed into an overall
knowledge graph of “gene-mutation-disease”
relations. 207 novel relations that were not included
in the ClinVar database were also discovered and
annotated in these linking articles, as well as
displayed in a distinct manner. These relations mostly
derive from the literature containing multiple entities
and complex relations, which made it difficult for
experts to extract these relations into ClinVar from
the linking articles based purely on reading and
comprehension.
3.2 Test Use Cases
3.2.1 Genetic Screening
Genetic screening, which mainly refers to
preimplantation genetic screening (PGS), helps lower
the risks of transmitting genetic defects to offspring,
implantation failure, and/or miscarriage during in
vitro fertilization (IVF) cycles (Lu et al., 2016).
Instead of designing and developing multiple probes
or primers for different disease testing, in this case the
clinician can obtain the entire set of potential diseases
on our platform by submitting the mutation calling
results from next-generation sequencing and
bioinformatics analysis.
Hypothetically, a subject is detected as a carrier of
both c.1118C>T and c.1320G>T mutations in the
CDH1 gene. Only after a tremendous effort in
consultation or experimentation can the clinician
begin to understand the pathogenicity of each
mutation in a traditional diagnostic manner. Our
platform, on the contrary, provides a more convenient
and efficient solution. The clinician can easily acquire
an initial impression that CDH1 c.1118C>T is likely
to correlate with hereditary diffuse gastric cancer
while CDH1 c.1320G>T plays a pathogenic role in
BIOINFORMATICS 2020 - 11th International Conference on Bioinformatics Models, Methods and Algorithms
56

Blepharocheilodontic syndrome (a rare autosomal
dominant condition), simply by conducting a search
for the CDH1 gene on our platform. Variant
properties can further support these results, and the
literature context can confirm them. In this case,
although both mutations are classified into missense
variants which alter amino acid sequences and affect
protein function, CDH1 c.1320G>T is causal to
Blepharocheilodontic syndrome (PMID: 28301459)
while CDH1 c.1118C>T is only weakly associated
with hereditary diffuse gastric cancer (PMID:
28492532). This evidence suggests that it is safe for
the clinician to conclude this subject is, in the future,
more likely to suffer from Blepharocheilodontic
syndrome than they are from hereditary diffuse
gastric cancer.
3.2.2 Differential Diagnosis
It is essential to characterize causative mutation in a
predisposing gene for the sake of differential
diagnosis among various hereditary syndromes with
their own distinctive organ-specific manifestations
that require different surveillance strategies (De Rosa
et al. 2015). Without costly and time-consuming
whole-exome analysis, or even whole-genome
analysis, the clinician can quickly and accurately
locate the pathogenicity mutations for a given disease
and confirm the classification of disease by means of
a few subsequent tailored tests.
For instance, a hypothetical patient was
previously diagnosed with hereditary breast and
ovarian cancer syndrome by traditional testing
methods (transvaginal ultrasonography and
mammography). Classifying the disease further
requires genetic variants information from
bioinformatics analysis. Unlike massive parallel
sequencing, target region sequencing is the purpose
of our platform. Indeed, target region sequencing
takes advantage of prior knowledge. Based on the
MeSH controlled vocabulary, hereditary breast and
ovarian cancer syndromes have four subtypes
including BROVCA1, BROVCA2, BROVCA3, and
BROVCA4. BROVCA1 is associated with 33
pathogenic mutations in BRCA1, while BROVCA2
is correlated with 13 pathogenic mutations in
BRCA2, and BROVCA3 is linked with 1 pathogenic
mutation in RAD51C. Consistent with the relation-
linked literature, the involving gene-targeted
premiers are then designed and applied for PCR. By
obtaining primer sequences, order of markers and
physical distances among D13S260, D13S1699,
D13S1698, D13S1697, D13S171, D13S1695 and
D13S1694 from the Ensembl Genome Browser
(PMID: 23929434), one can only identify and
confirm c.2808_2811del in BRCA2, suggesting that
the patient is most likely to suffer from breast-ovarian
cancer, familial 2 (BROVCA2).
4 DISCUSSION
4.1 Related Work
Instead of concentrating on bridging the gap between
distributed data sources, previous research has paid
more attention to knowledge base construction from
a heterogeneous collection of unstructured, semi-
structured and structured data or relation extraction
from biomedical literature.
We noted earlier that ClinVar, COSMIC, and
HGMD are commonly-used “gene-mutation-disease”
knowledge bases constructed that took months (or
longer) for skilled experts to construct. More
specifically, ClinVar is a submitter-driven repository
that archives reports of relationships among genomic
variants and phenotypes submitted by clinical
laboratories, researchers, clinicians, expert panels,
practice guidelines, and other groups or
organizations. COSMIC is a hand-curated resource
for exploring the effect of somatic mutations in
human cancer; it also offers detailed mutation data
along with additional information such as
environmental factors or patient pre-disposition.
HGMD is a comprehensive core collection of
germline mutations in nuclear genes that underlay
human inherited disease, functioning primarily
through combined electronic and manual search
procedures. Indeed, the constructed knowledge bases
achieve high quality and high acceptance thanks to
tremendous human effort, resources, and work hours,
but they fail to assure provenance tracking of the
curated knowledge, making themselves less
convincing and persuasive.
On the other hand, Singhal et al., Bravo et al. and
Ravikumar et al. worked on “gene-mutation-disease”
relation extraction tasks using text mining algorithms
and tools. Singhal et al. (2016) used a C4.5 decision
tree classifier to extract disease-gene-variant triplets
from all abstracts in PubMed that were related to a set
of important diseases. Bravo et al. (2015) used a
shallow linguistic kernel and dependency kernel to
identify gene-disease relationships from free text in
MEDLINE. Ravikumar et al. (2015) used a
dependency parse graph traversal algorithm to locate
mutation-disease associations within and across
sentences from MEDLINE abstracts. Having
achieved state-of-the-art performance in several
BROGUE: A Platform for Constructing and Visualizing “Gene-Mutation-Disease” Relation Knowledge Graphs to Support Biomedical
Research and Clinical Decisions
57

evaluations, these systems yet require model
optimization in classifier selection and feature
engineering, as well as knowledge representation and
subsequent knowledge integration like knowledge
fusion.
4.2 Limitations and Future Work
Good performance though our platform has achieved,
there still exists several limitations require further
improvement, which is database integration and
knowledge discovery.
According to a report of 2018 Molecular Biology
Database Collection in the journal Nucleic Acids
Research, there are up to 1,737 databases currently
available and publicly accessible online (Rigden and
Fernández, 2017). Built using an array of diverse
standards and for a number of different purposes,
these databases are difficult to unify without
meticulously designed and thoroughly thought-out
mapping models. We therefore temporarily selected
only one typical and authoritative database for each
element in our system (i.e. genes, mutation, disease,
relation, and literature) according to expert advice.
This lets us prove how our approach is both usable
and useful for platform construction, at the same time
giving us a chance to build a prototype of this
platform for constructing and visualizing “gene-
mutation-disease” relation knowledge graphs. Far
from adequate, the knowledge in our platform
demands supplementation and extension (e.g.
definition and synonym). In the future, we may resort
to crowdsourcing to establish multiple mapping
models for facilitating larger-scale data integration.
Serving a critical role in precision medicine, novel
“gene-mutation-disease” relation discovery from
biomedical literature remains under-studied. This
may be owed to a lack of fine-grained relation types
and high-quality labelled corpus, which together
make it difficult to construct state-of-the-art relation
extraction models. Indeed, even though processed
and curated by experts, the 207 newly-found relations
extracted from 527 articles are far from enough to
support biomedical research and clinical decision-
making in their own right. Recently, numerous deep
learning models have been applied in natural
language processing tasks, achieving strong
performance (Nguyen and Grishman, 2015; Lin et al.,
2016). In future work, we would design a task-
oriented “gene-mutation-disease” relation extraction
model using advanced deep neural networks like the
CNN or attention model, improving the overall
capability of detecting existing associations and
discovering new relations.
5 CONCLUSIONS
In this paper, we designed a platform development
methodology for biomedical knowledge graph
construction as well as visualization. Combining
several cutting-edge techniques and widely-used
tools, we integrated relevant information about
“gene-mutation-disease” relations from diverse data
sources and presented this information in various
manners to help users learn about their interested
entities or relations. Supported by integrated
knowledge and validated by application scenarios, we
have proven our platform capable of supporting
research conduction and decision making, but we
have also shown that it has great potential when it
comes to inclusive knowledge integration and
sustainable knowledge discovery.
ACKNOWLEDGEMENTS
This Research was funded by National Key R&D
Program of China (2016YFC0901900).
REFERENCES
Ashley, E. A. (2015). The precision medicine initiative: a
new national effort. Jama, 313(21), 2119-2120.
Beckmann, J. S., & Lew, D. (2016). Reconciling evidence-
based medicine and precision medicine in the era of big
data: challenges and opportunities. Genome medicine,
8(1), 134.
Bravo, À., Piñero, J., Queralt-Rosinach, N., Rautschka, M.,
& Furlong, L. I. (2015). Extraction of relations between
genes and diseases from text and large-scale data
analysis: implications for translational research. BMC
bioinformatics, 16(1), 55.
De Rosa, M., Pace, U., Rega, D., Costabile, V., Duraturo,
F., Izzo, P., & Delrio, P. (2015). Genetics, diagnosis and
management of colorectal cancer. Oncology reports,
34(3), 1087-1096.
Forbes, S. A., Beare, D., Gunasekaran, P., Leung, K.,
Bindal, N., Boutselakis, H., ... & Kok, C. Y. (2014).
COSMIC: exploring the world's knowledge of somatic
mutations in human cancer. Nucleic acids research,
43(D1), D805-D811.
Fox, G. N., & Moawad, N. S. (2003). UpToDate: a
comprehensive clinical database. Journal of family
practice, 52(9), 706-710.
Halavi, M., Maglott, D., Gorelenkov, V., & Rubinstein, W.
(2018). MedGen. In The NCBI Handbook [Internet].
2nd edition. National Center for Biotechnology
Information (US).
BIOINFORMATICS 2020 - 11th International Conference on Bioinformatics Models, Methods and Algorithms
58

Huslig, M. A., & Vardell, E. (2015). ClinicalKey 2.0:
Upgrades in a Point-of-Care Search Engine. Medical
reference services quarterly, 34(3), 343-352.
Jameson, J. L., & Longo, D. L. (2015). Precision
medicine—personalized, problematic, and promising.
Obstetrical & gynecological survey, 70(10), 612-614.
King, R. S. (2014). Visual storytelling with D3: an
introduction to data visualization in JavaScript.
Addison-Wesley Professional.
Kwon, D., Kim, S., Wei, C. H., Leaman, R., & Lu, Z.
(2018). ezTag: tagging biomedical concepts via
interactive learning. Nucleic acids research, 46(W1),
W523-W529.
Landrum, M. J., Lee, J. M., Riley, G. R., Jang, W.,
Rubinstein, W. S., Church, D. M., & Maglott, D. R.
(2013). ClinVar: public archive of relationships among
sequence variation and human phenotype. Nucleic
acids research, 42(D1), D980-D985.
Lin, Y., Shen, S., Liu, Z., Luan, H., & Sun, M. (2016,
August). Neural relation extraction with selective
attention over instances. In Proceedings of the 54th
Annual Meeting of the Association for Computational
Linguistics (Volume 1: Long Papers) (pp. 2124-2133).
Loper, E., & Bird, S. (2002, July). NLTK: the Natural
Language Toolkit. In Proceedings of the ACL-02
Workshop on Effective tools and methodologies for
teaching natural language processing and
computational linguistics-Volume 1 (pp. 63-70).
Association for Computational Linguistics.
Lu, L., Lv, B., Huang, K., Xue, Z., Zhu, X., & Fan, G.
(2016). Recent advances in preimplantation genetic
diagnosis and screening. Journal of assisted
reproduction and genetics, 33(9), 1129-1134.
Maglott, D., Ostell, J., Pruitt, K. D., & Tatusova, T. (2005).
Entrez Gene: gene-centered information at NCBI.
Nucleic acids research, 33(suppl_1), D54-D58.
McCartney, M., Treadwell, J., Maskrey, N., & Lehman, R.
(2016). Making evidence based medicine work for
individual patients. Bmj, 353, i2452.
Meyers, A. D. (2000). eMedicine Otolaryngology: an
online textbook for ENT specialists. Ear, nose & throat
journal, 79(4), 268-271.
Nguyen, T. H., & Grishman, R. (2015, June). Relation
extraction: Perspective from convolutional neural
networks. In Proceedings of the 1st Workshop on
Vector Space Modeling for Natural Language
Processing (pp. 39-48).
Ravikumar, K. E., Wagholikar, K. B., Li, D., Kocher, J. P.,
& Liu, H. (2015). Text mining facilitates database
curation-extraction of mutation-disease associations
from Bio-medical literature. BMC bioinformatics,
16(1), 185.
Rigden, D. J., & Fernández, X. M. (2017). The 2018
Nucleic Acids Research database issue and the online
molecular biology database collection. Nucleic acids
research, 46(D1), D1-D7.
Roberts, R. J. (2001). PubMed Central: The GenBank of the
published literature. Proceedings of the National
Academy of Sciences of the United States of America,
98(2), 381.
Saha, S. (2018, August). Open information extraction from
conjunctive sentences. In Proceedings of the 27th
International Conference on Computational Linguistics
(pp. 2288-2299).
Singhal, A., Simmons, M., & Lu, Z. (2016). Text mining
genotype-phenotype relationships from biomedical
literature for database curation and precision medicine.
PLoS computational biology, 12(11), e1005017.
Stenson, P. D., Mort, M., Ball, E. V., Evans, K., Hayden,
M., Heywood, S., ... & Cooper, D. N. (2017). The
Human Gene Mutation Database: towards a
comprehensive repository of inherited mutation data for
medical research, genetic diagnosis and next-
generation sequencing studies. Human genetics,
136(6), 665-677.
Vicknair, C., Macias, M., Zhao, Z., Nan, X., Chen, Y., &
Wilkins, D. (2010, April). A comparison of a graph
database and a relational database: a data provenance
perspective. In Proceedings of the 48th annual
Southeast regional conference (p. 42). ACM.
Wu, Y., Schuster, M., Chen, Z., Le, Q. V., Norouzi, M.,
Macherey, W., ... & Klingner, J. (2016). Google's
neural machine translation system: Bridging the gap
between human and machine translation. arXiv preprint
arXiv:1609.08144.
Zhao, D., Tong, F., & Luo, Z. (2019, February) Construct
Semantic Type of “Gene-mutation-disease” Relation by
Computer-aided Curation from Biomedical Literature.
In Proceedings of 10th International Conference on
Bioinformatics Models, Methods and Algorithms,
BIOINFORMATICS 2019-Part of 12th International
Joint Conference on Biomedical Engineering Systems
and Technologies, BIOSTEC 2019 (pp. 123-130).
BROGUE: A Platform for Constructing and Visualizing “Gene-Mutation-Disease” Relation Knowledge Graphs to Support Biomedical
Research and Clinical Decisions
59
