
Glioma Grade Classification via Omics Imaging
Lucia Maddalena
1 a
, Ilaria Granata
1 b
, Ichcha Manipur
1
, Mario Manzo
2 c
and Mario R. Guarracino
1 d
1
Inst. for High-Performance Computing and Networking, National Research Council, Via P. Castellino, 111, Naples, Italy
2
Information Technology Services, University of Naples “L’Orientale”, Via Nuova Marina, 59, Naples, Italy
Keywords:
Glioma Grade Classification, Metabolic Networks, Omics Imaging.
Abstract:
Omics imaging is an emerging interdisciplinary field concerned with the integration of data collected from
biomedical images and omics experiments. Bringing together information coming from different sources, it
permits to reveal hidden genotype-phenotype relationships, with the aim of better understanding the onset
and progression of many diseases, and identifying new diagnostic and prognostic biomarkers. In this work,
we present an omics imaging approach to the classification of different grades of gliomas, which are primary
brain tumors arising from glial cells, as this is of critical clinical importance for making decisions regarding
initial and subsequent treatment strategies. Imaging data come from analyses available in The Cancer Imag-
ing Archive, while omics attributes are extracted by integrating metabolic models with transcriptomic data
available from the Genomic Data Commons portal. We investigate the results of feature selection for the two
types of data separately, as well as for the integrated data, providing hints on the most distinctive ones that
can be exploited as biomarkers for glioma grading. Moreover, we show how the integrated data can provide
additional clinical information as compared to the two types of data separately, leading to higher performance.
We believe our results can be valuable to clinical tests in practice.
1 INTRODUCTION
Gliomas are neuroepithelial tumors affecting glial
cells of the central nervous system (CNS) and are the
most common primary form of brain tumors. The
classification and grading of gliomas have evolved
over time, since 1926 until the modern classifica-
tion based on the World Health Organization (WHO)
classification of CNS tumors, first published in 1979
and then revised several times, most recently in
2016 (Louis et al., 2016). The WHO classifica-
tion system categorizes gliomas from grade I (lowest
grade) to grade IV (highest grade), based on several
morphologic, histological, and molecular attributes.
The differentiation between low-grade gliomas
(LGGs) and high-grade gliomas (HGGs) is critical,
since the prognosis, and thus the therapeutic strategy,
could differ substantially depending on the grade. In-
deed, HGGs are usually treated with surgical resec-
tion, followed by radiation therapy and chemother-
a
https://orcid.org/0000-0002-0567-4624
b
https://orcid.org/0000-0002-3450-4667
c
https://orcid.org/0000-0001-8727-9865
d
https://orcid.org/0000-0003-2870-8134
apy. If they are misdiagnosed as LGGs, then they
will be treated less effectively (Togao et al., 2016).
A further distinction has to be made in the context
of HGGs, where the last grade, namely GlioBlas-
toma Multiforme (GBM), is the most aggressive dif-
fuse heterogeneous glioma with poor prognosis and
complex treatment, so much to be considered almost
untreatable (Soeda et al., 2015).
The extraction of large amounts of quantitative
features from medical images, often referred to as ra-
diomics (Lambin et al., 2012; Kumar et al., 2012),
has emerged in the last decade as a powerful method-
ology to quantify the characteristics of tumors in a
non-invasive manner, providing imaging biomarkers
for supporting clinical decision-making (Cho et al.,
2018). This approach has been investigated also for
the classification of glioma grades (Law et al., 2003;
Zacharaki et al., 2009; Togao et al., 2016; Cho et al.,
2018; Ertosun and Rubin, 2015; Khawaldeh et al.,
2018), where conventional and advanced Magnetic
Resonance Imaging (MRI) techniques are adopted to
extract a variety of imaging features.
With the advent of high-throughput arrays and
Next Generation Sequencing technologies, it is
now possible to integrate information coming from
82
Maddalena, L., Granata, I., Manipur, I., Manzo, M. and Guarracino, M.
Glioma Grade Classification via Omics Imaging.
DOI: 10.5220/0009167700820092
In Proceedings of the 13th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2020) - Volume 2: BIOIMAGING, pages 82-92
ISBN: 978-989-758-398-8; ISSN: 2184-4305
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

biomedical images with the omics data, i.e., those ob-
tained by the large-scale characterization and quan-
tification of molecules present in cells. A new in-
terdisciplinary field is emerging, concerned with the
integration of data collected from biomedical images
and omics analyses. Often named imaging genomics
(Hariri and Weinberger, 2003; Jaffe, 2012; Lee et al.,
2017) or radiogenomics (Acharya et al., 2018; Gillies
et al., 2016; Sala et al., 2017), we refer to it as
omics imaging (Antonelli et al., 2019), to better re-
flect the wide variety of omics (genomics, transcrip-
tomics, proteomics, other omics) data that can be ex-
ploited and combined with structural, functional, and
molecular imaging data. Bringing together informa-
tion coming from different sources, it permits to re-
veal hidden genotype-phenotype relationships, with
the aim of better understanding the onset and progres-
sion of various diseases, and identifying new diagnos-
tic and prognostic biomarkers (Ranjbar and Mitchell,
2017).
Several omics imaging studies investigated the re-
lationship between GBM imaging phenotypes, eval-
uated by MR images (Diehn et al., 2008; Zinn
et al., 2011; Beig et al., 2017) or CT images (Jain
et al., 2012), and gene-expression profiles, assessed
by cDNA microarray (Diehn et al., 2008; Zinn et al.,
2011; Smedley and Hsu, 2018) or RNA-seq analy-
ses (Beig et al., 2017). Some of them also investigated
the possible predictive value of the omics imaging ap-
proach for the survival of GBM patients (e.g., (Diehn
et al., 2008; Gevaert et al., 2014)). However, to our
knowledge, none of them was devoted to the classifi-
cation of the glioma grades.
In this work, we present an omics imaging ap-
proach to the classification of glioma grades. We start
integrating a dataset with both imaging and omics
data for each patient, collecting the data from publicly
available sources. Imaging features extracted by MRI
data come from publicly available analyses (Bakas
et al., 2017a), while, based on previous work (Granata
et al., 2019), we show how to integrate omics vari-
ables to span a new feature space. Finally, we re-
port and analyze results using or not feature selection
and oversampling, empirically proving that the inte-
gration of imaging and omics features, together with
these techniques, can lead to improved performance.
2 MATERIALS AND METHODS
2.1 Data
Omics data come from The Cancer Genome Atlas
(TCGA, tcga-data.nci.nih.gov), a multi-institutional
comprehensive collection of various molecularly
characterized tumor types, whose data are avail-
able through the NCI’s Genomic Data Commons
portal (gdc.cancer.gov). Specifically, we exploited
FPKM (fragments per kilobase per million reads
mapped) normalized gene counts from RNA sequenc-
ing data of two brain cancer projects, TCGA-GBM
and TCGA-LGG. TCGA-GBM contains data for 161
samples, while TCGA-LGG contains data for 511
samples (see the “O” column in Table 1). For these
Table 1: Number of samples for the Omics dataset (O), the
Imaging dataset (I), and their intersection (OI).
Collection O I OI
TCGA-GBM 161 135 30
TCGA-LGG 511 108 104
Total 672 243 134
data, we extracted features based on a metabolic
model of brain tissue (Agren et al., 2012), down-
loaded from the Metabolic Atlas database (metabol-
icatlas.org). Specifically, by combining samples
of gene expression data with the tissue-specific
metabolic model, a weighted directed multigraph for
each sample is obtained (Granata et al., 2019). Its
nodes represent the involved metabolites; the edges
connect interacting metabolites, and their weights are
the expression values of the enzymes catalyzing the
reactions in which the couple of metabolites is in-
volved. The multigraphs are reduced to simple graphs
as in (Granata et al., 2018), by taking the mean of the
expression values for multiple enzymes connecting
two nodes in the same reaction and then summing up
the means of different reactions connecting the couple
of metabolites. This way, each network contains 8458
edges, reduced to 1375 by eliminating edges having
weights common to all samples, used in the follow-
ing as omics features (O). The scheme of the proce-
dure for collecting omics features is reported in Fig.
1 (left).
Imaging data consist of multimodal MRI im-
ages publicly available from The Cancer Imag-
ing Archive (TCIA, cancerimagingarchive.net) (Clark
et al., 2013), where case-linked diagnostic pre-
surgical images are available for a large subset of the
TCGA genomically analyzed cases. Imaging features
are those available in the TCIA archive as analysis re-
sults provided by Bakas et al. (Bakas et al., 2017a;
Bakas et al., 2017b; Bakas et al., 2017c), extracted
from images of the TCGA-GBM and TGCA-LGG
collections. A subset of the available radiological
data was selected by the authors, including those that
refer to pre-operative baseline scans with available
MRI modalities of at least T1-weighted pre-contrast
Glioma Grade Classification via Omics Imaging
83

Figure 1: Omics and Imaging data. Omics data (left) are
obtained by the simplification of multigraphs constructed
by the metabolic model of brain tissue used for the RNA
sequencing data. Imaging data (right) consist of radiomics
features volumetrically extracted by MRI images.
(T1), T1-weighted post-contrast (T1-Gd), T2, and
T2-FLAIR. After pre-processing and segmentation of
different glioma sub-regions, a panel of more than
700 features was extracted volumetrically by the se-
lected image data. These features provide quantita-
tive information regarding intensity, volumes, mor-
phology, histogram-based, and textural parameters, as
well as spatial information and parameters extracted
from glioma growth models. The features have been
computed by the authors for 135 TCGA-GBM sub-
jects and 108 TGCA-LGG subjects (see “I” column
in Table 1). Among them, those for 102 GBM and 65
LGG samples are publicly available through TCIA,
while the remaining features, used for the MICCAI
Brain Tumor Segmentation 2018 (BraTS 2018)
1
test
data set, are available upon demand.
Matching the subject IDs of imaging and omics
data, we obtained a total of 30 samples from TCGA-
GBM and 104 samples from TCGA-LGG having both
omics and imaging data (see “OI” column in Table 1).
In the following, the whole sets of omics, imaging,
and omics imaging features for the matched samples
will be denoted as O, I, and OI, respectively.
In case of feature selection (see Sect. 2.2.1), two
integration strategies will be considered: 1) the inte-
grated data consists of the features selected among the
whole OI concatenated set (again referred to as OI),
and 2) the integrated data consists of the concatena-
tion of the two sets of selected omics and imaging
features (referred to as OpI).
2.2 Classification Procedure
The procedure adopted for classification follows a
typical workflow, as reported in Fig. 2. The input
1
https://www.med.upenn.edu/sbia/brats2018.html
data (either O, I, or OI features) is partitioned into a
training and a testing set containing 66% and 33% of
the samples, respectively. The partitioning is chosen
randomly, taking care to have the same distribution of
samples over classes, both in training and in test set.
The training set is used to determine an optimal subset
of features (see Sect. 2.2.1). This subset is extracted
from both the training and the testing set and used for
classification (see Sect. 2.2.2).
Figure 2: Scheme of the classification procedure. Data is
partitioned into a training and a testing set. The training
set is used to determine an optimal subset of features. This
subset is extracted from both the training and the testing set
and used for classification.
2.2.1 Feature Selection
It is well-known that feature selection is generally a
fundamental step prior to classification (Wang et al.,
2016). This is even more compelling in the case of
omics imaging. Indeed, first of all, the overall number
of features is in the order of tens of thousand, giving
rise to the well-known curse of dimensionality. The
associated mathematical problem is ill-posed because
the number of variables is generally far larger than
the number of samples. When the number of sam-
ples is low, the features might appear highly corre-
lated. Moreover, the number of features represent-
ing the two types of data is very unbalanced: omics
data are described by tens of thousands of variables,
whereas the imaging variables are usually in the order
of hundreds. This imbalance produces a high proba-
bility of selecting features by chance from the omics
data. Finally, the information-to-noise ratio is differ-
ent in imaging and omics data, which might lead to
overfitting classification models.
For feature selection, we use SVM-RFE (Guyon
et al., 2002), a wrapper method based on recursive
feature elimination using Support Vector Machines
(SVM) (Vapnik, 1995), that provides a ranked list
BIOIMAGING 2020 - 7th International Conference on Bioimaging
84
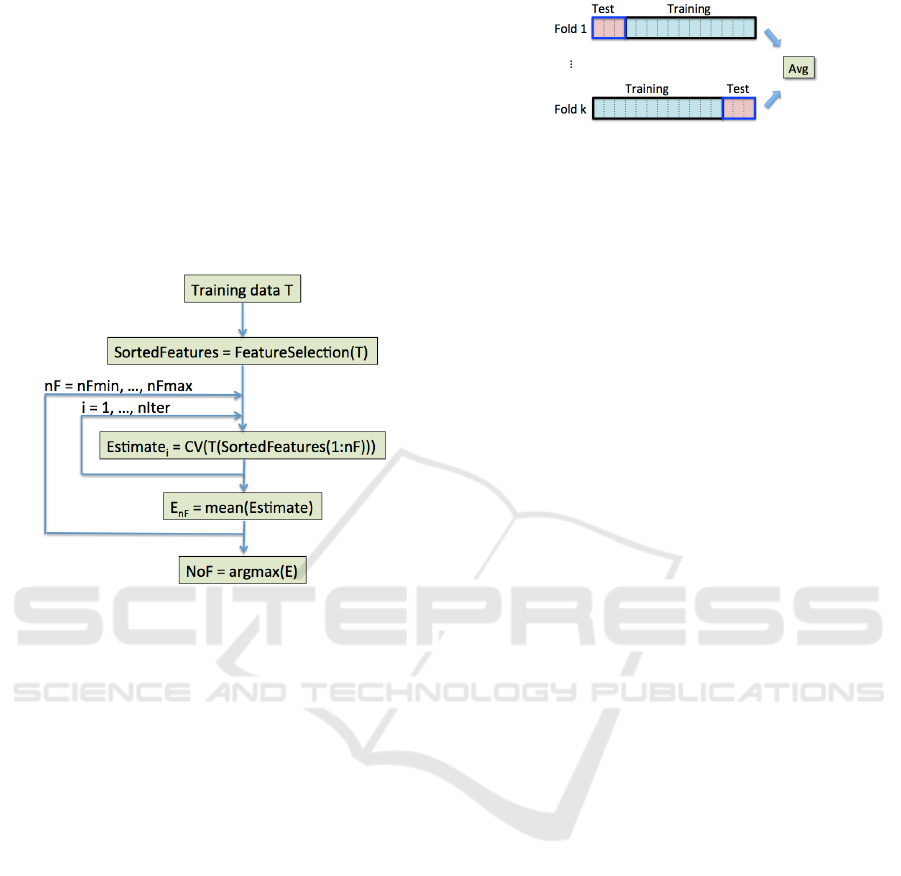
of features ordered according to their relevance. To
choose the optimal number of features to be selected
from a dataset, we perform a k-fold cross-validation
(CV) of a classification model (see Sect. 2.2.2)
trained using the first NoF sorted features of the train-
ing data and choose the value of NoF that maximizes
the achieved performance across niter iterations of
the process (see Fig. 3). In the experiments, we set
k=5 and niter=10; NoF is searched in the interval
[10,20], experimentally proved to be suitable for all
O, I, and OI data, maximizing the AUC measure.
Figure 3: Selection of NoF features. Once features from
the training data are ranked, their optimal number NoF is
chosen as the one that maximizes average performance, es-
timated through CV.
2.2.2 Classification
For supervised classification, as well as for choos-
ing the optimal number of selected features, several
classifiers can be considered. In the experiments, we
report results using the k-Nearest Neighbor classifier
(k=1), deemed to be among the top 10 techniques for
data mining (Wu et al., 2007).
2.2.3 Evaluation
For evaluating the classification results, the classifica-
tion procedure of Fig. 2 is repeated a number numIter
of times (in the experiments, numIter is set to 50),
each time randomly permuting the data and validat-
ing the classification of the extracted features through
k-fold CV (see Fig. 4). In the experiments, k is set to
5. Overall performance values are obtained as the av-
erage of those obtained at each iteration (i.e., for each
of the partitions).
Figure 4: Scheme of k-fold cross-validation. Data is divided
into k subsets. At each of the k iterations, one subset is
used as the test set and the other k-1 subsets are used as the
training set. The k results are averaged to produce a single
estimation.
2.2.4 Handling Imbalanced Data
Considering a binary classification problem, a dataset
is said to be imbalanced if there exists an under-
represented concept (a minority class) when com-
pared to the other (a majority class). The imbalance
ratio (IR), i.e., the ratio of the cardinalities of the ma-
jority and the minority classes, is usually adopted to
establish the imbalance severity. Prediction models
built from imbalanced datasets are often biased to-
wards the majority concept, which is especially crit-
ical when there is a higher cost of misclassifying the
minority examples, as in our case for diagnosing the
highest glioma grade. For recent discussions on han-
dling imbalanced data, see (Peeken et al., 2018; San-
tos et al., 2018; Manzo, 2019). The problem is even
more challenging in the context of omics imaging,
since imaging and omics techniques analyze phenom-
ena that might have intrinsic differences and different
sensitivity levels (Antonelli et al., 2019).
As the IR in our datasets is quite high (IR=3.47),
in our experiments, we adopted the Adaptive syn-
thetic sampling approach for imbalanced learning
(ADASYN) oversampling method (Haibo He et al.,
2008), an extension of Synthetic Minority Over-
sampling Technique (SMOTE) (Chawla et al., 2002)
that synthetically creates new samples of the minority
class via linear interpolation between existing minor-
ity class samples. Those minority samples that are
harder to learn are given greater importance; thus,
they are oversampled more often. Therefore, most of
the new samples lay in the vicinity of the boundary
between the two classes, rather than in the interior of
the minority class.
2.2.5 Cross Validation for Imbalanced Data
In our framework, CV is adopted for both feature se-
lection (see Sect. 2.2.1) and overall evaluation (see
Sect. 2.2.3). It is well known that the joint appli-
cation of CV with oversampling should be handled
with care (Santos et al., 2018). Specifically, over-
sampling should not be applied to the entire original
Glioma Grade Classification via Omics Imaging
85

data, over which to perform CV and model evaluation,
as this procedure would lead to building biased mod-
els and producing overoptimistic error estimates. In-
stead, oversampling should be applied during CV (see
Fig. 5), only on the training data of each fold, so that
exact or similar replicas of a given pattern produced
by oversampling cannot be found in both the training
and test sets. In our experiments using oversampling,
Figure 5: Scheme of k-fold cross-validation with oversam-
pling. At each step, oversampling (for balancing green and
red classes) is performed only on subsets used for training.
Red samples with * indicate artificial samples added to the
minority (red) class.
the standard CV (as depicted in Fig. 4) is replaced by
the CV with oversampling sketched in Fig. 5, for both
feature selection and evaluation.
3 RESULTS AND DISCUSSION
3.1 Performance Measures
In our experiments, we consider several performance
metrics, defined in terms of the number of true pos-
itives (TP), true negatives (TN), false positives (FP)
and false negatives (FN)
• Accuracy (Acc): measures the percentage of cor-
rectly classified examples and is computed as
Acc =
T P + T N
T P + FN + FP + T N
; (1)
• Specificity (Spec): measures the percentage of
negative examples correctly identified and is de-
fined as
Spec =
T N
T N + FP
; (2)
• Sensitivity (Sens): also referred to as Recall, it
measures the percentage of positive examples cor-
rectly classified and is computed as
Sens =
T P
T P + FN
; (3)
• Precision (Prec): corresponds to the percentage
of positive examples correctly classified, consid-
ering the set of all the examples classified as pos-
itive, and is defined as
Prec =
T P
T P + FP
; (4)
• F-measure (F
β
): shows the compromise between
sensitivity and precision, obtained as
F
β
= (1 + β
2
) ·
Prec · Sens
(β
2
· Prec) + S ens
, (5)
where β ∈ R
+
weights the role of sensitivity and
precision. In our experiments, we consider F
1
(i.e., β=1);
• Adjusted F-measure (AGF): addresses imbal-
anced data, giving more weight to patterns
correctly classified in the minority (positive)
class (Maratea et al., 2014). It is defined as
AGF =
√
F
2
· InvF
0.5
, (6)
where F
2
is computed as in Eq. (5) for β=2 and
InvF
0.5
is computed for β=0.5 and through an in-
version of the confusion matrix, where positive
samples become negative and vice versa;
• G-mean (Gm): represents the geometric mean of
the accuracy of both classes and is defined as
Gm =
√
Sens · Spec; (7)
• Area Under the ROC Curve (AUC): makes use
of the Receiver Operating Characteristics (ROC)
curve to exhibit the trade-off between the classi-
fier’s TP and FP rates (He and Garcia, 2009).
For all the above metrics, higher values indicate better
performance results. Besides completeness, the rea-
son for using so many metrics, rather than only the
frequently adopted Acc, is that, in the case of un-
balanced datasets, Acc is biased towards the major-
ity class (He and Garcia, 2009). In the experiments,
the majority class (LGG) is assumed as the negative
class, while the minority class (GBM) is assumed as
the positive class.
3.2 Performance Analysis
Performance results on the testing sets are reported in
Table 2. Here, we compare the results obtained with-
out feature selection (“All features”) with those ob-
tained applying feature selection (“Selected features”)
as well as those obtained using oversampling (right)
with those obtained without oversampling (left). The
sets of features considered are only imaging features
(I), only omics features (O), and omics imaging fea-
tures (OI). In the case of feature selection, we further
consider the second integration strategy described in
Sect. 2.1, i.e., the set of omics imaging features (OpI)
obtained by concatenating, for each patient, the I and
O features separately selected (i.e., feature selection
is not performed on OI, but only separately on I and
O).
BIOIMAGING 2020 - 7th International Conference on Bioimaging
86
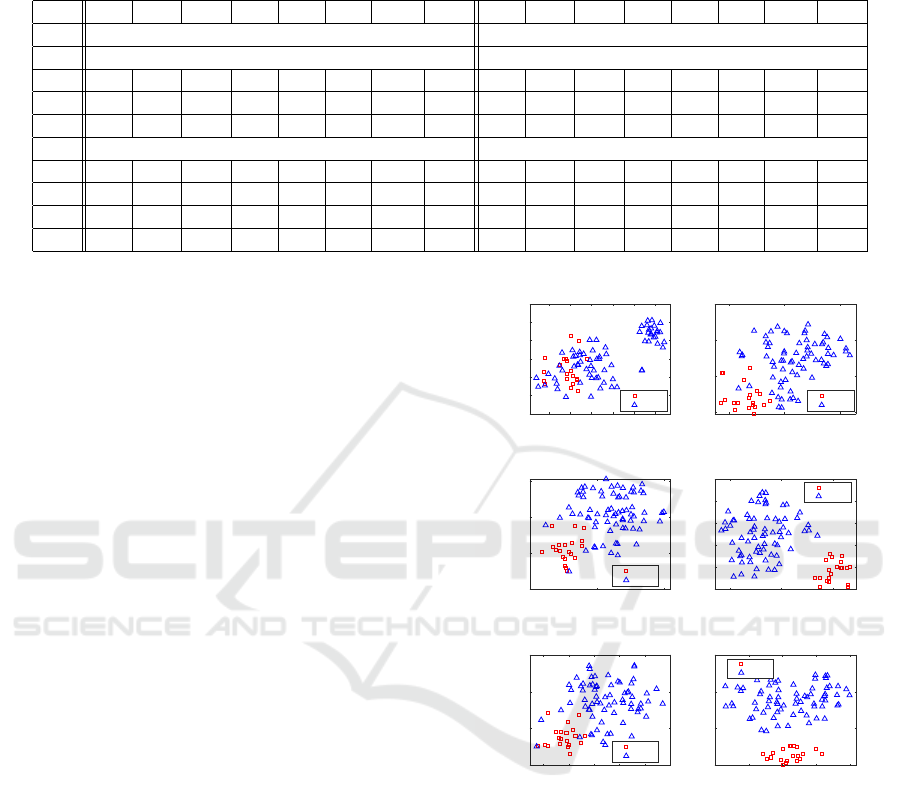
Table 2: Testing performance (%) with/without feature selection (All features/Selected features), as well as with/without
oversampling, on the I, O, and OI features. In case of feature selection, we also consider the omics imaging features (OpI)
obtained by concatenating the I and O features separately selected.
Data Acc Sens Spec Prec F
1
Gm AUC AGF Acc Sens Spec Prec F
1
Gm AUC AGF
Testing WITH oversampling Testing WITHOUT oversampling
All features All features
I 67.3 87.0 61.5 40.8 54.7 70.2 74.2 75.2 72.7 82.0 69.9 46.2 57.7 72.2 76.0 75.5
O 92.1 93.0 91.9 81.7 84.9 91.8 92.5 92.5 91.2 70.0 97.4 90.0 75.8 79.7 83.7 79.7
OI 89.8 98.0 87.4 74.3 82.7 92.2 92.7 93.5 93.3 90.0 94.2 86.3 85.9 91.3 92.1 91.7
Selected features Selected features
I 80.5 69.5 83.7 58.2 60.3 70.6 76.6 71.7 81.2 60.4 87.3 58.2 56.3 64.9 73.9 65.5
O 95.1 90.7 96.5 90.8 88.9 92.2 93.6 92.5 94.3 84.9 97.1 90.3 85.4 88.6 91.0 88.7
OI 95.0 91.1 96.1 90.2 88.8 92.4 93.6 92.7 94.6 86.0 97.1 90.5 86.2 89.3 91.5 89.5
OpI 92.8 90.1 93.7 85.3 85.2 90.6 91.9 91.2 92.4 82.0 95.5 86.4 81.4 86.0 88.7 86.3
From Table 2, it can be observed that feature se-
lection almost always leads to a substantial improve-
ment according to any performance measure. Excep-
tions can be observed for Sensitivity in testing with
oversampling (bottom-left of Table 2), which is re-
duced from 87%, 93%, and 98% to 69.5%, 90.7%,
and 91.1% for the I, O, and OI features, respectively.
Consequently, also AGF is reduced, as it weighs more
the correct classification of minority samples. How-
ever, these performance reductions are compensated
in terms of augmented values for all the other met-
rics. Instead, in case of results obtained without over-
sampling (right part of Table 2), we can observe more
cases where performance decreases when using fea-
ture selection, especially for the imaging features.
The beneficial role of feature selection is con-
firmed by the visual analysis based on T-distributed
Stochastic Neighbor Embedding (t-SNE), reported
in Fig. 6. t-SNE is a nonlinear dimensional-
ity reduction technique that allows embedding of
high-dimensional data for visualization in a low-
dimensional space (van der Maaten and Hinton,
2008). It models each high-dimensional sample by
a two- or three-dimensional point in such a way that
similar samples are modeled by nearby points and dis-
similar samples are modeled by distant points with
high probability. It is capable of retaining the local
structure of the high-dimensional data, while also re-
vealing some important global structure, such as the
presence of clusters at several scales. Figs. 6-(a)-(c)
provide visual representations of the glioma data used
for training mapped into the 2D Euclidean space by t-
SNE, considering both all the features (left plots) and
the selected features (right plots). Data are colored
to reflect the ground truth classification (red squares
for GBM and blue triangles for LGG). In addition
to showing these scatterplots, we also display a met-
ric called neighborhood hit (NH) (Paulovich et al.,
2008). For a given number of neighbors k (in our ex-
-4 -2 0 2 4 6
All features
-10
-8
-6
-4
-2
0
2
t-SNE (NH = 0.785185)
GBM
LGG
-5 0 5
Selected features
-5
0
5
10
t-SNE (NH = 0.912963)
GBM
LGG
(a)
-5 0 5
All features
-10
-5
0
5
t-SNE (NH = 0.916667)
GBM
LGG
-5 0 5
Selected features
-6
-4
-2
0
2
4
t-SNE (NH = 0.994444)
GBM
LGG
(b)
-6 -4 -2 0 2 4
All features
-10
-5
0
5
t-SNE (NH = 0.911111)
GBM
LGG
-4 -2 0 2 4
Selected features
-10
-5
0
5
t-SNE (NH = 1.000000)
GBM
LGG
(c)
Figure 6: Role of feature selection: t-SNE of (a) I, (b) O,
and (c) OI data considering all the features (left plots) or
the selected features (right plots) for training data, where
feature selection is performed.
periments, k=6), the NH for a projected point p ∈ R
2
is defined as the ratio of its k-nearest neighbors (ex-
cept p itself) that belong to the same class as the cor-
responding observation. The NH for a projection is
defined as the average NH over all its points. Intu-
itively, a high NH corresponds to a projection where
the real classes (ground truth) are visually well sep-
arated. Therefore, as suggested in (Rauber et al.,
2018), the NH metric provides a good quantitative
characterization of a t-SNE projection. Fig. 6-(a) re-
ports the t-SNE visual representations for one of the
Glioma Grade Classification via Omics Imaging
87

random partitions of the imaging data. Here, we ob-
serve that the two classes do not appear well sepa-
rated when using all the features (left plots). Instead,
using the subset of selected features (right plots) al-
lows to better group data belonging to the same class
in periphery areas of the 2D plane, even though the
groups are still not spatially separated. The gain in us-
ing feature selection is well reflected by the increase
in the NH value. From the visual representations for
the omics data reported in Fig. 6-(b), we observe that
the two classes appear better separated than for the
imaging data, also when using all the features. Using
the subset of selected features leads to data clusters
that are much more spatially separated, also showing
much higher NH values. Similar observations can be
done for the omics imaging data, whose t-SNE visual
representations are reported in Fig. 6-(c), showing
that the subset of selected features leads to correctly
cluster the mapped data in separate plane regions in a
way that is consistent with their classification and the
NH values when using only the selected features are
extremely high.
The above analysis based on both performance
metrics and visual representations by t-SNE high-
lights the higher discriminating power of omics imag-
ing data, especially if coupled with feature selection
and oversampling, as compared to any of the types of
data separately.
3.2.1 Comparisons with Other Methods
To complete our analysis and discussion of the pro-
posed framework, we wish to compare our results
with those obtained by different approaches. How-
ever, due to the novelty of our omics imaging ap-
proach, to the best of our knowledge, no other method
exists that has been evaluated on the same dataset
and that can be compared for glioma grade classifi-
cation. Therefore, in Table 3, we summarize the per-
formance of several methods for classifying glioma
grades, bearing in mind that each has been validated
by its authors on a different set of data.
The method in (Law et al., 2003) uses relative
cerebral blood volume (rCBV) measurements derived
from perfusion MRI and metabolite ratios from pro-
ton MR spectroscopy, with a cohort of 160 patients.
Table 3 reports sensitivity and specificity for discrim-
inating the 120 HGG and 40 LGG samples (computed
by the authors for correct identification of HGGs,
i.e., assuming HGG as positive) achieved both using
solely rCBV and combining rCBV with metabolite ra-
tios (m.r.).
The work in (Togao et al., 2016) investigates the
diagnostic performance of intravoxel incoherent mo-
tion imaging for glioma grade classification using
Table 3: Performance (%) of methods for glioma grades
classification.
Method Acc Sens Spec AUC
Law2003-rCBV 95.0 57.5
Law2003-rCBV+m. r. 93.3 60.0
Togao2016 96.6 81.2 95.0
Zacharaki2009 87.8 84.6 95.5 89.6
Cho2018 88.5 95.1 70.2 90.3
Ertosun2015 96.0 98.0 94.0
Khawaldeh2018 91.3 87.5 95.3
I 80.5 69.5 83.7 76.6
O 95.1 90.7 96.5 93.6
OI 95.0 91.1 96.1 93.6
several parameters, with a cohort of 45 patients. Ta-
ble 3 reports their best sensitivity, specificity, and
AUC values for discriminating the 29 HGG and 16
LGG samples, obtained based on the volume fraction
within a voxel of water flowing in perfused capillars.
The method in (Zacharaki et al., 2009) is based
on a total of 161 imaging features, including shape,
intensity, and texture features, extracted by different
MRI modalities on a set of 74 brain tumors. Fea-
ture selection is obtained using a SVM-based recur-
sive feature elimination process similar to the one we
adopted, but using a leave-one-out CV. Table 3 reports
the accuracy, sensitivity, specificity, and AUC values
achieved by weighted SVM to differentiate between
the 22 LGG and 52 HGG samples.
Cho et al. (Cho et al., 2018) quantify gliomas
with a radiomics approach and use the results to dif-
ferentiate between 210 GBM and 75 LGG samples.
They consider data from the MICCAI Brain Tumor
Segmentation 2017 Challenge (Menze et al., 2015),
derived from the TCGA-GBM and TGCA-LGG col-
lections. A set of 468 quantitative radiomics features
(based on shape, histogram, texture, and intensity)
is computed from four MRI modalities, considering
three glioma sub-regions. The minimum redundancy
maximum relevance algorithm is adopted to select
five features, used to build three different classifier
models. Table 3 reports the average accuracy, sen-
sitivity, specificity, and AUC values achieved by the
classifiers in the testing cohort as defined in the BraTS
challenge.
Two deep learning-based methods have recently
been proposed for classifying glioma grades. In (Er-
tosun and Rubin, 2015), a deep learning-based clas-
sification pipeline is proposed using digital pathology
images of whole tissue slides obtained by TCGA. Ta-
ble 3 reports the accuracy, sensitivity, and specificity
values obtained by the authors to differentiate be-
tween the 52 LGG and 48 GBM microbiopsy samples
randomly selected from independent test slides. Deep
BIOIMAGING 2020 - 7th International Conference on Bioimaging
88

learning-based classification for grading of glioma tu-
mors is also considered in (Khawaldeh et al., 2018).
However, the model is trained using single 2D slices
of FLAIR MRI images available in TCIA. The label-
ing available for each patient was extended to label,
for each MRI, a subset of slices showing the lesion.
Table 3 reports the accuracy, sensitivity, and speci-
ficity values computed by the confusion matrix pub-
lished by the authors for a (more difficult) three-class
classification problem on 587 samples (213 GBM,
235 LGG, and 139 healthy).
For direct comparison, in Table 3, we also re-
port the best performance results for the proposed ap-
proach using feature selection and oversampling on I,
O, and OI features. Overall, we can conclude that the
proposed approach based on using OI features shows
performance similar to or higher than all the com-
pared methods in terms of all the considered perfor-
mance metrics.
3.3 Analysis of Selected Features
Table 4 reports the most frequently selected features
from I, O, and OI data, respectively. These have been
collected, for each dataset, as those selected in the
training set of each of the 50 iterations of the eval-
uation procedure. The numbers on the left indicate
the number of times (over 50) each feature has been
selected. Among the 704 imaging features (named ac-
cording to the nomenclature provided by the authors
(Bakas et al., 2017b; Bakas et al., 2017c)), only 193
of them have been selected at least once when pro-
cessing imaging data. The most frequently selected
concern volume, histogram, intensity, spatial, and tex-
tural properties of the MRI images. Among the 1375
omics features (named according to the acronyms of
the involved metabolites), only 153 of them have been
selected at least once when processing omics data.
Only 131 of the 2079 omics imaging features have
been selected at least once when processing omics
imaging data. Among them, the most frequently se-
lected imaging features concern volume and spatial
properties, while the most frequently selected omics
features are also those most frequently chosen while
applying feature selection to O alone.
The strategy of integrating omics data into
metabolic models is proper to systems biology ap-
proach, where the integration of multiple informa-
tion coming from different data sources is exploited
to investigate the links among molecular components
of varying nature. Here we used the gene expres-
sion data and the gene-protein-reaction relationships
(GPR) to weight the connections among metabolites
consumed and produced in enzymes-catalyzed reac-
tions. Through this approach, we can get insights
into the metabolism even starting from gene-level
data. Indeed, the extracted features are metabolites
connections, and thus, we can recover useful infor-
mation from single metabolites, whole reactions, be-
longing subsystems, and enzymes, providing a wider
choice of candidate biomarkers. Furthermore, focus-
ing our attention on metabolites, we aim at overcom-
ing the limit imposed by gene expression quantifica-
tion, which requires the extraction of the RNA from
the brain tissue. Indeed, it is possible to measure the
metabolites abundance through LC-MS/MS-based
quantification in both cerebrospinal fluid and brain
(Fuertig et al., 2016), through in-vivo proton magnetic
resonance spectroscopy (1H-MRS) (Jansen et al.,
2006). The first three mostly recurrent features ob-
tained from omics and omics imaging are exactly the
same, confirming their strong discriminative power.
In particular, the first two are selected in almost all
the iterations, and the third one in half of them. All
features highlight an increased nutrient request in
more aggressive cancers, made of cells that prolifer-
ate and invade more rapidly. Indeed, the first metabo-
lite couple (m00247c m00569c), 1,3-bisphospho-D-
glycerate → 2,3-bisphospho-D-glycerate, is part of
the Glycolysis/Gluconeogenesis subsystem, and the
associated reaction is catalyzed by PGAM1. The lat-
ter is the brain isoform of the phosphoglyceric acid
mutase, a well-known enzyme in cancer research,
since it is involved in the so-called Warburg effect,
the aerobic glycolisis that provides a selective ad-
vantage to cancer cells for growing and proliferat-
ing. Its abundance is highly correlated to aggres-
siveness and poor prognosis of tumors (Sun et al.,
2018). The second omics and omics imaging feature
(m02579c m02579s) is the transport of ammonium
ions NH4+ from cytoplasm to extracellular compart-
ment. Several studies have associated ammonium
ions to growth, death, and regulation of apoptotic pro-
cesses, depending on cell types. It is generally rec-
ognized as a waste product of glutaminolysis, a fun-
damental energy source for proliferating cells, and is
deleterious to cells; that is the reason for finding the
transport toward the outside of cells. Surely NH4+
abundance and transport are correlated to the rate of
energy production (Abusneina and Gauthier, 2016).
The third metabolites link (m01972l m01430l) in-
volves the following reaction: glucosylceramide pool
+ H2O → ceramide pool + glucose. It is part of the
glycosphingolipid metabolism in lysosomes, which
are lipids particularly abundant in the nervous system,
involved in many biological processes. The impair-
ment of their production or catabolism leads to differ-
ent lysosome storage diseases, which are a hallmark
Glioma Grade Classification via Omics Imaging
89
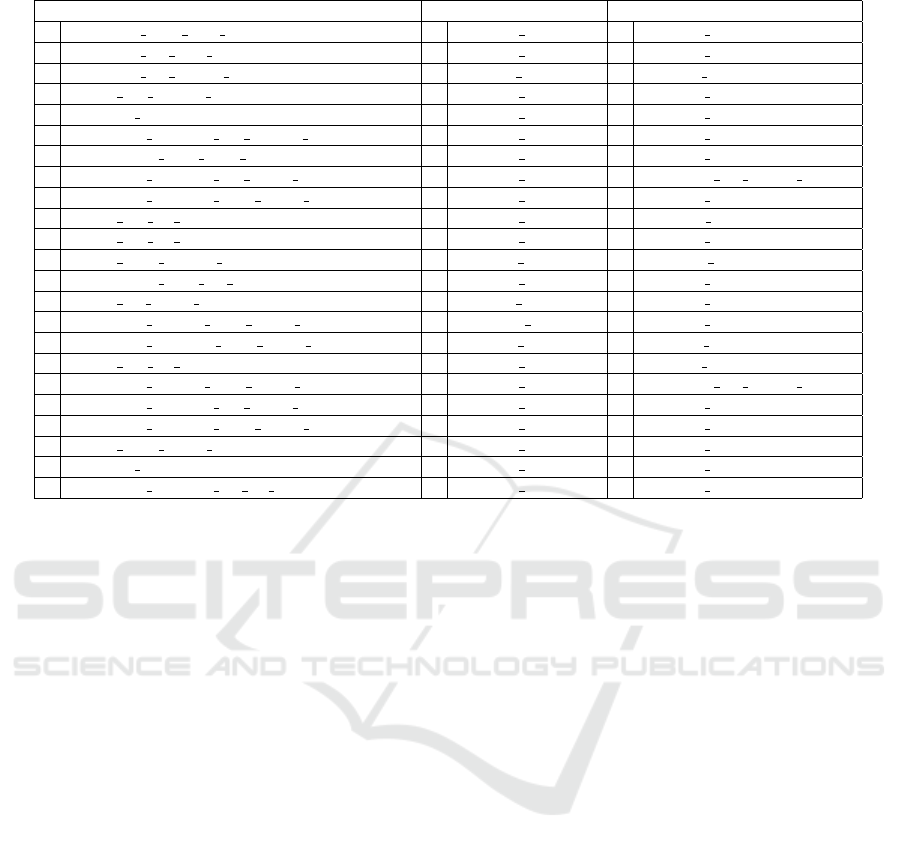
Table 4: Most frequently selected imaging, omics, and omics imaging features, with their frequency.
Imaging Omics Omics&Imaging
36 VOLUME NET over TC 49 m00247c m00569c 47 m00247c m00569c
32 VOLUME ET over TC 47 m02579c m02579s 43 m02579c m02579s
30 VOLUME ET OVER WT 24 m01972l m01430l 28 m01972l m01430l
26 HISTO ED FLAIR Bin10 23 m01990c m01992c 24 m01688c m01680c
25 SPATIAL Temporal 21 m02133c m02471c 23 m02344c m02344s
25 TEXTURE GLSZM ED FLAIR SZHGE 20 m02344c m02344s 19 m01990c m01992c
23 INTENSITY STD NET T1Gd 19 m01688c m01680c 18 m02133c m02471c
21 TEXTURE GLSZM ED T1Gd LGZE 18 m02845c m02806c 12 VOLUME ET OVER WT
21 TEXTURE GLSZM NET T1Gd SZHGE 15 m01307c m02335c 11 m00809c m00968c
18 HISTO ED T1 Bin3 13 m01972c m01430c 11 m01868g m01869g
16 HISTO ED T1 Bin2 11 m01939c m00247c 11 m02658c m02812c
16 HISTO NET FLAIR Bin10 10 m01913s m01910s 10 SPATIAL Insula
15 INTENSITY STD ET T1Gd 9 m01307c m02818c 10 m01115c m02471c
14 HISTO ET T1Gd Bin7 9 m02008l m01910l 10 m01939c m00247c
14 TEXTURE GLCM NET T1Gd AutoCorrelation 8 m00826m m02189m 10 m01965c m01965g
14 TEXTURE GLRLM NET T1Gd SRLGE 8 m01821s m01822s 9 m01913s m01910s
13 HISTO ED T1 Bin6 8 m01965c m01965g 9 m02008l m01910l
13 TEXTURE GLCM NET T1Gd SumAverage 8 m02658c m02812c 7 VOLUME ET OVER BRAIN
12 TEXTURE GLSZM ED T1Gd SZLGE 7 m00351c m00349c 7 m00554c m00555c
10 TEXTURE GLSZM NET T1Gd SZE 7 m00809c m00968c 7 m01307c m02335c
9 HISTO NET T1Gd Bin10 7 m01968c m01968r 7 m01673c m01755c
9 SPATIAL Insula 6 m00554c m00555c 7 m01690c m01939c
9 TEXTURE GLSZM ET T2 SZHGE 6 m01690c m01939c 7 m02845c m02806c
of neuronopathic forms of the disease (Boomkamp
and Butters, 2008; Russo et al., 2018).
4 CONCLUSIONS
The availability of data from both omics and imaging
experiments for each patient provides deeper insight
into the classification of disease states, as in the case
of brain tumors. In this work, we analyze in detail
the problem of integrating publicly available data to
discriminate between brain tumor subtypes. Imaging
features come from analyses available in the TCIA
archive, while, in the case of transcriptomic data, fea-
tures are extracted in terms of metabolic information
through the integration of gene expression values into
a genome-wide metabolic model of the brain tissue.
This approach allows us to get a broader range of in-
formation, going from the enzymes to the metabo-
lites and the associated reactions. Focusing on the
metabolic alterations is a widespread strategy in can-
cer research, both because the transformed cells adopt
an energy/metabolic reprogramming (Jeon and Hay,
2018) and because the metabolites and the enzymes
represent good targets for diagnostic and therapeu-
tic challenges (Luengo et al., 2017). The adopted
framework takes into account several strategies that
can lead to better and fairer results, including feature
selection and data balancing, and their correct incor-
poration in the entire procedure. We show that the
integration of omics and imaging data, also thanks to
these precautions, can provide more accurate results
than their separate use, even using a small number
of significant features from both the types of exper-
iments.
Future research will be devoted to both new meth-
ods and data. Indeed, our implementation choices
have been guided by the established literature, rather
than by their specific suitability for the problem at
hand. It will be interesting to further investigate
whether the adoption of different classifier models,
data balancing strategies or feature selection algo-
rithms specifically devised for integrating multimodal
data, may lead to even better results for this or sim-
ilar applications. Moreover, we will try to integrate
larger volumes of data coming from different experi-
ments and to generalize our findings to more massive
datasets of various diseases. This means, on the one
hand, producing imaging features with standard and
FAIR procedures, like those adopted in this study. On
the other hand, this leads our attention to other types
of omics data (e.g., those coming from the blood)
that could help reducing invasive interventions. This
would facilitate and enhance the role of omics imag-
ing studies as a support to the medical doctors.
BIOIMAGING 2020 - 7th International Conference on Bioimaging
90

ACKNOWLEDGEMENTS
The work was carried out also within the activities
of M.R. Guarracino and L. Maddalena as members
of the INdAM Research group GNCS. M. Manzo ac-
knowledges the guidance and supervision of Prof. Al-
fredo Petrosino during the years spent working to-
gether. The authors would like to thank G. Trerotola
for the technical support.
REFERENCES
Abusneina, A. and Gauthier, E. R. (2016). Ammonium ions
improve the survival of glutamine-starved hybridoma
cells. Cell & bioscience, 6(1):23.
Acharya, U. R., Hagiwara, Y., Sudarshan, V. K., et al.
(2018). Towards precision medicine: from quantita-
tive imaging to radiomics. J. Zhejiang Univ. Sci. B,
19(1):6–24.
Agren, R., Bordel, S., Mardinoglu, A., et al. (2012). Recon-
struction of genome-scale active metabolic networks
for 69 human cell types and 16 cancer types using
INIT. PLoS computational biology, 8(5):e1002518.
Antonelli, L., Guarracino, M. R., Maddalena, L., et al.
(2019). Integrating imaging and omics data: A review.
Biomed Signal Process Control, 52:264–280.
Bakas, S., Akbari, H., Sotiras, A., et al. (2017a). Advancing
The Cancer Genome Atlas glioma MRI collections
with expert segmentation labels and radiomic features.
Scientific data, 4.
Bakas, S., Akbari, H., Sotiras, A., et al. (2017b). Seg-
mentation labels and radiomic features for the pre-
operative scans of the TCGA-GBM collection. The
Cancer Imaging Archive.
Bakas, S., Akbari, H., Sotiras, A., et al. (2017c). Seg-
mentation labels and radiomic features for the pre-
operative scans of the TCGA-LGG collection. The
Cancer Imaging Archive.
Beig, N. et al. (2017). Radiogenomic analysis of hypoxia
pathway reveals computerized MRI descriptors pre-
dictive of overall survival in glioblastoma. In Proc.
SPIE, volume 10134, pages 101341U–101341U–10.
Boomkamp, S. D. and Butters, T. D. (2008). Glycosphin-
golipid disorders of the brain. In Lipids in Health and
Disease, pages 441–467. Springer.
Chawla, N. V., Bowyer, K. W., Hall, L. O., et al. (2002).
Smote: Synthetic minority over-sampling technique.
J. Artif. Int. Res., 16(1):321–357.
Cho, H.-h., Lee, S.-h., Kim, J., et al. (2018). Classification
of the glioma grading using radiomics analysis. PeerJ,
6.
Clark, K. et al. (2013). The Cancer Imaging Archive
(TCIA): Maintaining and operating a public infor-
mation repository.
Journal of Digital Imaging
,
26(6):1045–1057.
Diehn, M. et al. (2008). Identification of noninvasive imag-
ing surrogates for brain tumor gene-expression mod-
ules. Proc. of the National Academy of Sciences of the
United States of America, 105(13):5213–5218.
Ertosun, M. G. and Rubin, D. L. (2015). Automated grad-
ing of gliomas using deep learning in digital pathol-
ogy images: A modular approach with ensemble of
convolutional neural networks. In AMIA 2015 Annual
Symposium Proc., pages 1899–1908.
Fuertig, R., Ceci, A., Camus, S. M., Bezard, E., Luippold,
A. H., and Hengerer, B. (2016). LC–MS/MS-based
quantification of kynurenine metabolites, tryptophan,
monoamines and neopterin in plasma, cerebrospinal
fluid and brain. Bioanalysis, 8(18):1903–1917.
Gevaert, O., Mitchell, L., Achrol, A., et al. (2014).
Glioblastoma multiforme: exploratory radiogenomic
analysis by using quantitative image features. Radiol-
ogy, 273(1):168–74.
Gillies, R. J., Kinahan, P. E., and Hricak, H. (2016). Ra-
diomics: Images are more than pictures, they are data.
Radiology, 278(2):563–577. PMID: 26579733.
Granata, I., Guarracino, M. R., Kalyagin, V. A., et al.
(2018). Supervised classification of metabolic net-
works. In IEEE Int. Conf. on Bioinformatics and
Biomedicine (BIBM), Madrid, Spain, December 3-6,
2018, pages 2688–2693.
Granata, I., Guarracino, M. R., Kalyagin, V. A., et al.
(2019). Model simplification for supervised classifi-
cation of metabolic networks. Annals of Mathematics
and Artificial Intelligence.
Guyon, I., Weston, J., Barnhill, S., et al. (2002). Gene se-
lection for cancer classification using support vector
machines. Machine Learning, 46(1):389–422.
Haibo He, Yang Bai, Garcia, E. A., et al. (2008). ADASYN:
Adaptive synthetic sampling approach for imbalanced
learning. In 2008 IEEE Int. Joint Conf. on Neural Net-
works (IEEE World Congress on Computational Intel-
ligence), pages 1322–1328.
Hariri, A. R. and Weinberger, D. R. (2003). Imaging ge-
nomics. British Medical Bulletin, 65(1):259–270.
He, H. and Garcia, E. A. (2009). Learning from imbalanced
data. IEEE T Knowl Data En, 21(9):1263–1284.
Jaffe, C. C. (2012). Imaging and genomics: Is there a syn-
ergy? Radiology, 264(2):329–331.
Jain, R., Poisson, L., Narang, J., Scarpace, L., et al. (2012).
Correlation of perfusion parameters with genes related
to angiogenesis regulation in glioblastoma: a feasibil-
ity study. AJNR Am J Neuroradiol., 33(7):1343–8.
Jansen, J. F., Backes, W. H., Nicolay, K., and Kooi, M. E.
(2006). 1h MR spectroscopy of the brain: absolute
quantification of metabolites. Radiology, 240(2):318–
332.
Jeon, S.-M. and Hay, N. (2018). Expanding the concepts of
cancer metabolism.
Khawaldeh, S., Pervaiz, U., Rafiq, A., et al. (2018). Non-
invasive grading of glioma tumor using magnetic res-
onance imaging with convolutional neural networks.
Applied Sciences, 8(1).
Kumar, V. et al. (2012). Radiomics: the process
and the challenges. Magnetic Resonance Imaging,
30(9):1234–1248. Quantitative Imaging in Cancer.
Glioma Grade Classification via Omics Imaging
91

Lambin, P., Rios-Velazquez, E., Leijenaar, R., et al. (2012).
Radiomics: Extracting more information from medi-
cal images using advanced feature analysis. European
Journal of Cancer, 48(4):441–446.
Law, M., Yang, S., and Wang, H. a. (2003). Glioma grading:
Sensitivity, specificity, and predictive values of perfu-
sion MR imaging and proton MR spectroscopic imag-
ing compared with conventional MR imaging. Amer-
ican Journal of Neuroradiology, 24(10):1989–1998.
Lee, G., Lee, H. Y., Ko, E. S., et al. (2017). Radiomics
and imaging genomics in precision medicine. Precis
Future Med, 1(1):10–31.
Louis, D. N., Perry, A., Reifenberger, G., Von Deimling,
A., Figarella-Branger, D., Cavenee, W. K., Ohgaki,
H., Wiestler, O. D., Kleihues, P., and Ellison, D. W.
(2016). The 2016 World Health Organization clas-
sification of tumors of the central nervous system: a
summary. Acta neuropathologica, 131(6):803–820.
Luengo, A., Gui, D. Y., and Vander Heiden, M. G. (2017).
Targeting metabolism for cancer therapy. Cell chemi-
cal biology, 24(9):1161–1180.
Manzo, M. (2019). Kgearsrg: Kernel graph embedding on
attributed relational sift-based regions graph. Machine
Learning and Knowledge Extraction, 1(3):962–973.
Maratea, A., Petrosino, A., and Manzo, M. (2014). Ad-
justed f-measure and kernel scaling for imbalanced
data learning. Information Sciences, 257:331–341.
Menze, B. H., Jakab, A., Bauer, S., et al. (2015). The mul-
timodal brain tumor image segmentation benchmark
(brats). IEEE Trans Med Imaging, 34(10):1993–2024.
Paulovich, F. V., Nonato, L. G., Minghim, R., et al. (2008).
Least square projection: A fast high-precision mul-
tidimensional projection technique and its application
to document mapping. IEEE Trans Vis Comput Graph,
14(3):564–575.
Peeken, J. C., Bernhofer, M., Wiestler, B., et al. (2018).
Radiomics in radiooncology - challenging the medical
physicist. Physica Medica, 48:27–36.
Ranjbar, S. and Mitchell, J. R. (2017). Chapter 8 - an intro-
duction to radiomics: An evolving cornerstone of pre-
cision medicine. In Depeursinge, A., Al-Kadi, O. S.,
and Mitchell, J., editors, Biomedical Texture Analysis,
pages 223 – 245. Academic Press.
Rauber, P. E., Falcao, A. X., and Telea, A. C. (2018). Pro-
jections as visual aids for classification system design.
Information Visualization, 17(4):282–305.
Russo, D., Della Ragione, F., Rizzo, R., Sugiyama, E.,
Scalabr
`
ı, F., Hori, K., Capasso, S., Sticco, L., Fior-
iniello, S., De Gregorio, R., et al. (2018). Glycosph-
ingolipid metabolic reprogramming drives neural dif-
ferentiation. The EMBO journal, 37(7):e97674.
Sala, E. et al. (2017). Unravelling tumour heterogene-
ity using next-generation imaging: Radiomics, ra-
diogenomics, and habitat imaging. Clin. Radiol.,
72(1):3–10.
Santos, M. S., Soares, J. P., Abreu, P. H., et al. (2018).
Cross-validation for imbalanced datasets: Avoiding
overoptimistic and overfitting approaches [research
frontier]. IEEE Comp. Int. Mag., 13(4):59–76.
Smedley, N. F. and Hsu, W. (2018). Using deep neural net-
works for radiogenomic analysis. In 2018 IEEE 15th
Int. Symposium on Biomedical Imaging (ISBI 2018),
pages 1529–1533.
Soeda, A., Hara, A., Kunisada, T., Yoshimura, S.-i., Iwama,
T., and Park, D. M. (2015). The evidence of glioblas-
toma heterogeneity. Scientific reports, 5:7979.
Sun, Q., Li, S., Wang, Y., Peng, H., Zhang, X., Zheng, Y.,
Li, C., Li, L., Chen, R., Chen, X., et al. (2018). Phos-
phoglyceric acid mutase-1 contributes to oncogenic
mTOR-mediated tumor growth and confers non-small
cell lung cancer patients with poor prognosis. Cell
Death & Differentiation, 25(6):1160.
Togao, O., Hiwatashi, A., Yamashita, K., et al. (2016).
Differentiation of high-grade and low-grade diffuse
gliomas by intravoxel incoherent motion MR imaging.
Neuro-Oncology, 18(1):132–141.
van der Maaten, L. and Hinton, G. (2008). Visualizing data
using t-SNE. Journal of Machine Learning Research,
9:2579–2605.
Vapnik, V. (1995). The Nature of Statistical Learning The-
ory. Springer-Verlag.
Wang, L., Wang, Y., and Chang, Q. (2016). Feature se-
lection methods for big data bioinformatics: A survey
from the search perspective. Methods, 111:21–31. Big
Data Bioinformatics.
Wu, X., Kumar, V., Ross Quinlan, J., et al. (2007). Top 10
algorithms in data mining. Knowl. Inf. Syst., 14(1):1–
37.
Zacharaki, E. I., Wang, S., Chawla, S., et al. (2009). Classi-
fication of brain tumor type and grade using MRI tex-
ture and shape in a machine learning scheme. Magn
Reson Med, 62:a609–1618.
Zinn, P., Majadan, B., et al. (2011). Radiogenomic map-
ping of edema/cellular invasion MRI-phenotypes in
glioblastoma multiforme. PLoS ONE, 6(10).
BIOIMAGING 2020 - 7th International Conference on Bioimaging
92
