
Finding Potential Inhibitors of COVID-19
Angela Kralevska
1
, Marija Velichkovska
1
, Viktor Cicimov
1
, Tome Eftimov
1,2
and
Monika Simjanoska
1
1
Ss. Cyril and Methodius University, Faculty of Computer Science and Engineering, Skopje, North Macedonia
2
Computer Systems Department, Jo
ˇ
zef Stefan Institute, Ljubljana, Slovenia
monika.simjanoska@finki.ukim.mk
Keywords:
COVID-19, Sars-CoV-2, Treatment, Drug, Inhibitors, Molecular Docking, Virtual Screening.
Abstract:
COVID-19 is an infectious disease caused by virus SARS-CoV-2 that spread globally due to its high conta-
gious nature and became an ongoing pandemic. The lack of vaccines and drugs to treat infected patients is a
great problem in the fight against this pandemic. Molecular docking is one of the best approaches to search for
potential drugs in real time with possibilities to apply at COVID-19. In this experiment, molecular docking
studies of fourteen ligands were carried out with three important proteins of SARS-CoV-2, i.e. main protease,
ACE2, and spike glycoprotein. From the obtained results, we observed that many of the tested molecules
showed better dock score in comparison to remdesivir and dexamethasone, drugs that are claimed to be ef-
fective against COVID-19. Combining the dock score and other properties, we believe that auranetin can be
further explored for potential use against COVID-19.
1 INTRODUCTION
Year 2020 has brought along with itself a global
tragedy in a form of pandemic named COVID-19. It
has proven to be a highly pathogenic and transmit-
table viral infection causing the severe acute respi-
ratory syndrome. Started in Wuhan, China, it has
rapidly spread throughout the world. SARS-CoV-
2 has caused around 50 million infections and more
than one million deaths (Yamin, 2020).
Despite the worsening trends of COVID-19, large-
scale studies report that no drugs are validated to have
significant efficiency in clinical treatment of patients
diagnosed with COVID-19. So far, remdesivir has
been approved by the Food and Drug Administration
Agency, only for the treatment of COVID-19 patients
that require hospitalization (FDA, 2020). The world
at the moment is in a dire need for new drugs against
COVID-19, ones that combine efficiency with mini-
mal side effects, but also are inexpensive and readily
available.
In the fight against coronavirus, scientists have
come up with three strategies for developing new
drugs. The first strategy is testing existing broad-
spectrum anti-virals. This category encompass inter-
ferons, ribavirin, and cyclophilin inhibitors used to
treat coronavirus pneumonia. Using existing molec-
ular databases to screen for molecules that may have
therapeutic effect on coronavirus is the second strat-
egy, and the third strategy is based on the pathological
characteristics and genomic information of different
coronaviruses with the aim to develop new targeted
drugs from scratch. Theoretically, the drugs found
through these approaches would exhibit better anti-
coronavirus effects, however, the research procedure
might last for more than 10 years (Wu et al., 2020).
For the development of medicines for treating
SARS-CoV-2, the fastest way is to find potential
molecules from the marketed drugs. Remdesivir is
considered the most promising antiviral agent. It
works by inhibiting the activity of RNA-dependent
RNA polymerase to stop the virus from reproducing
and making copies of itself. RdRp inhibitor favipi-
ravir is also being clinically evaluated for its efficacy
at treating COVID-19 patients. Lopinavir/ritonavir,
the protease inhibitor, plus ribavirin were shown to
be effective against SARS-CoV in vitro. Hydroxy-
chloroquine plus azithromycin is another promising
alternative, that showed excellent clinical efficacy on
Chinese patients against COVID-19. Many other in-
hibitors, such as monoclonal and polyclonal antibod-
ies and teicoplanin, which inhibits the viral genome
exposure in cytoplasm, are under investigation for the
treatment of SARS-CoV-2 (Jean et al., 2020).
Corticosteroid called dexamethasone was the first
shown to reduce Covid-19 deaths. A study of more
110
Kralevska, A., Velichkovska, M., Cicimov, V., Eftimov, T. and Simjanoska, M.
Finding Potential Inhibitors of COVID-19.
DOI: 10.5220/0010246901100117
In Proceedings of the 14th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2021) - Volume 3: BIOINFORMATICS, pages 110-117
ISBN: 978-989-758-490-9
Copyright
c
2021 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

than 6,000 people found that dexamethasone reduced
deaths by one-third in patients on ventilators, and by
one-fifth in patients on oxygen (Peter et al., 2020).
This drug was recommended for people hospitalized
with COVID-19 who are on mechanical ventilators or
need supplemental oxygen by the U.S. National Insti-
tutes of Health. If dexamethasone and other corticos-
teroids are given for less severe COVID-19 infection,
they may be harmful (Sparks, 2020).
Disrupting this virus’s self-replication machinery
could be one of the ideal targets without causing any
harm to the host. SARS-CoV-2 has an active site for
the inhibitors. The spikes are responsible for the at-
tachment of the viruses on the surface and their sub-
sequent entry into the host cells. One of the major
causes behind the virus infecting multiple hosts is be-
cause of its loosely bound receptor-binding domains
(Singh et al., 2020).
In this research, we are exploring some ligands
with the aim to determine the level up to which they
inhibit main protease, ACE2 and spike glycoprotein.
The rest of the paper is organized as follows. We
review related work in Section 2. The methods are
presented in Section 3. In Section 4 is explained the
process of retrieving and preprocessing the data for
this project. The whole process of performing molec-
ular docking is briefly explained in Section 5. The
results are reported in Section 6, followed by the con-
clusions from this study in Section 7.
2 RELATED WORK
The molecular docking has been actively researched
to find potential drugs that can be used for COVID-
19 (Narkhede et al., 2020). In (Omar et al., 2020),
the authors used molecular docking and showed that
Quercetin, Hispidulin, Cirsimaritin, Sulfasalazine,
Artemisin and Curcumin showed better potential in-
hibition than Hydroxy-Chloroquine against COVID-
19 main protease active site. Another study was
focused on molecular docking of 18 ligands with
three three therapeutic target proteins of SARS-CoV-
2, i.e. RNA-dependent RNA polymerase (RdRp),
angiotensin-converting enzyme 2 (ACE2) and spike
glycoprotein (SGp), where phytochemicals has bet-
ter dock score compared to the paracetmol and hy-
droxychloroquine (Vardhan and Sahoo, 2020). Rib-
avirin, remdesivir, chloroquine and luteolin have been
also studied, where it was shown that luteolin bind
with a high affinity to the same sites of the main pro-
tease of SARS-CoV-2 as the control molecule (Yu
et al., 2020). Finding potential inhibitors for SARS-
CoV-2 main protease (Mpro) using a combination of
molecular docking and fast pulling of ligand (FPL)
simulations has been also researched in (Pham et al.,
2020). It was shown that 20 compounds were able
to bind well to SARS-CoV-2 Mpro, among them
five top are: are periandrin V, penimocycline, cis-
p-Coumaroylcorosolic acid, glycyrrhizin, and ural-
saponin B.
3 METHODS
The problem with the coronavirus can be viewed as a
typical chemical problem while we are trying to ob-
tain potential medicines that will work as inhibitors
in the process of transcription. Computer-aided drug
design (CADD) is a rational drug design technology,
which enables drug discovery based on knowledge
of target structures, functional properties and mech-
anisms. When the target protein structure is known,
structure-based approaches, such as molecular dock-
ing, can be used (Hung and Chen, 2014).
The path to drug discovery is long and chal-
lenging. This process starts with the discovery of
molecules that show efficacy in a simple screen,
called hits. The interaction between two molecules
can happen in a form of interaction of a protein and
protein, or, a protein and small molecule. Molecular
docking helps in predicting the intermolecular frame-
work and suggest the binding modes responsible for
inhibition of the protein (Aaftaab et al., 2019).
There are two basic components which distinguish
the variety of docking softwares available to choose
from. These are the sampling algorithm and scoring
function. After doing some research we have found
that most appropriate software for our research would
be AutoDock Vina since it is newly designed and im-
proved version of the AutoDock program. This ver-
sion adopted a new knowledge-based scoring function
with a Monte Carlo sampling technique and the Broy-
den–Fletcher–Goldfarb–Shanno (BFGS) method for
local optimization (Nataraj et al., 2017).
AutoDock Vina (Oleg and Arthur, 2010) tends to
be much faster than AutoDock, and can take advan-
tage of multiple CPUs or CPU cores to significantly
shorten its running time.
The first step in performing docking studies is to
find the appropriate compounds that would be fetched
from PubChem. There are many compounds to be
chosen from in PubChem, since it is the world’s
largest free chemistry database (PubChem, 2020),
however, we will pay attention to the ones that have
not been yet explored for COVID-19 in the previous
researches. Also, we will rely on the information that
the spike glycoprothein (Huang et al., 2020), main
Finding Potential Inhibitors of COVID-19
111
protease (Ullrich and Nitsche, 2020), or ACE2 (Ghe-
blawi et al., 2020) are regions that can be used to in-
hibit the transcription of the virus. Each compound
will be docked and depending on the affinity that is
produced, the one with highest rank will be chosen as
a candidate. Those compounds that form a bond and
as a result have higher energy, will be chosen.
Virtual screening based on molecular docking will
be used in order to explore all compounds that are able
to bind with the proteins and inhibit the virus.
4 DATA AND PREPROCESSING
In the process of molecular docking, the first step is
to obtain proteins and ligands from online databases.
Next step is preprocessing the molecules and trans-
forming them to the format needed for the process of
molecular docking.
4.1 Proteins and Ligands Selection
To study the protein-ligand interactions, we retrieved
proteins in ‘.pdb’ format from RSCB protein data
bank (RCSB, 2020). We downloaded the crystallo-
graphic 3D protein structures of main protease with
PDB ID: 6LU7, spike glycoprotein with PDB ID:
2GHV and ACE2 (Angiotensin Converting Enzyme
2) with PDB ID: 1R42.
The ligands were chosen after performing a thor-
ough literature research (Kouznetsova et al., 2020).
Also, we used the similarity structure search option
that is offered by the PubChem database. Compo-
nents with similar structure to ones that have already
shown significant docking affinity with the proteins
were taken into consideration.
Therefore, we decided to use the following lig-
ands in our research: Sinensetin, Alnetin, Au-
ranetin, Hesperetin, Nobiletin, Obacunone, Pedalitin,
Pomiferin, Tangeretin, Eucalyptin, Citromycetin, Tri-
fluoperazine, Beta D Mannose and Dimetylsulphox-
ide. The affinity of these components will be also
compared to the affinity of Remdesivir and Dex-
amethasone that are already used as treatment for
COVID-19.
4.2 Protein Preparation
The protein structures we are using are crystal struc-
tures complexes with ligand(s). Therefore, to dock
the desired ligand with the protein in that particular
position we need to remove the bound ligand by re-
moving hetatoms from the PDB file. If the docking
with our ligand is done without removing the already
complexed ligand, the obtained results will be incor-
rect. During the preprocessing step, except removing
hetatoms, water molecules were deleted, and we opti-
mized hydrogen bonds structures.
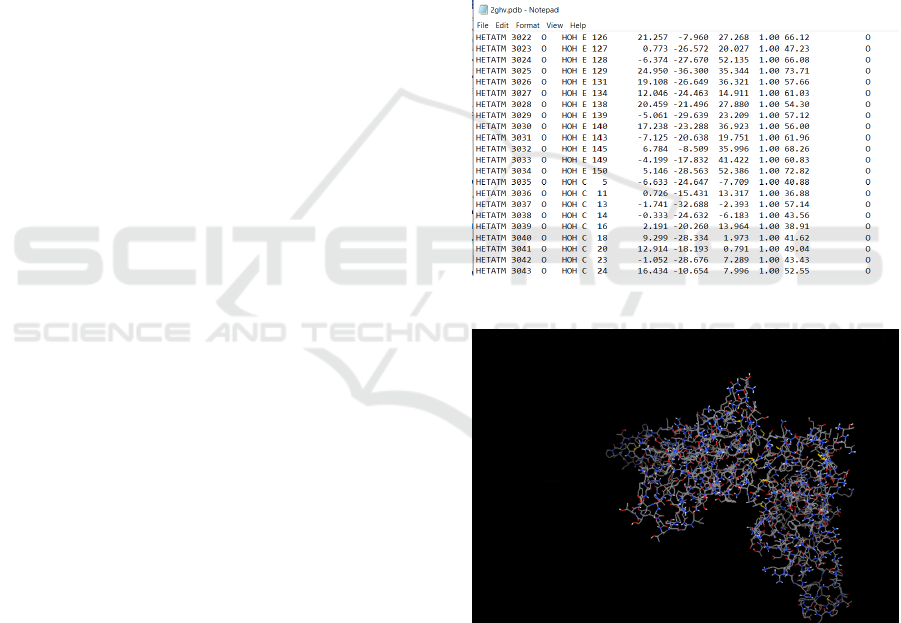
Fig. 1 depicts ‘2ghv.pdb’ file, i.e. the spike glyco-
protein. It can be noticed that the hetatoms are bind-
ing with only two chains: Chain E and Chain C. These
are the only chains we will need for docking. The
preprocessing is done with MGLTools (MGLTools,
2020) software developed at the Molecular Graphics
Laboratory (MGL) and AutoDock Tools.
As the final preprocessing step, the files are trans-
formed from ‘.pdb’ to a ‘.pdbqt’ format that is re-
quired for docking in AutoDock Vina. The look of
the molecule after preprocessing is shown in Fig. 2.
Figure 1: Spike protein hetatoms binding chains.
Figure 2: Spike protein after preprocessing.
4.3 Ligand Preparation
Ligands were taken in ‘.sdf’ format directly from the
PubChem Database (National Library of Medicine),
and converted into ‘.pdb’ format using the PyMOL
software (PyMOL, 2020). After that, with AutoDock
tools the files were transformed to ’.pdbqt’. Example
of prepared ligand is shown in Fig. 3.
BIOINFORMATICS 2021 - 12th International Conference on Bioinformatics Models, Methods and Algorithms
112

Figure 3: Ligand hispidulin after preprocessing.
5 MOLECULAR DOCKING
5.1 Defining Binding Site
There are two ways in which docking can be done:
specific site docking or blind docking. In blind dock-
ing, because the binding sites are unknown, the whole
protein is used. For specific site docking, expert
chemistry knowledge is needed to define the binding
site in the protein. Information about the place where
other ligands are binded is necessary, because another
ligand is suposed to be binded in the same position.
If the amino acids are known, the region we are inter-
ested at can be found with AutoDock tools. There-
fore, since we have the information describing the
docking sites, we are going to do specific site dock-
ing.
Targeted docking sites for ACE2 are (375, 505,
273, 345, 371, 30 to 41, 82 to 84, and 353 to 357). For
molecular docking of the main protease, the selected
cavity is the binding site of inhibitor N3. Binding sites
for the spike glycoprotein in chain E are (361 to 368,
391 to 399, 401, 414, 416, 420, 422, 489, 490 and
494), and in chain C they are (361 to 368, 391 to 395,
397 to 401, 414, 416, 420, 422, 489, 490 and 494).
Figure 4: Defining binding site of main protease with
AutoDock Tools.
Fig. 4 presents an example of main protease with
selected amino acids. They are represented with yel-
low crosses on the picture and it can be noticed that
all of them are in particular part of the molecule. That
part should be taken into consideration for the process
of docking.
5.2 Grid Box for Docking
Next step is enclosing the binding site for the ligand
in a grid box. We used AutoDock tools software to
position the grid box because there we can select the
binding site amino acids and place the box at the most
appropriate location of the protein. Spike glycopro-
tein with selected amino acids, enclosed in a grid box
is shown on Fig 5. This way we defined the following
boxes.
Grid box of size (74, 70, 80) centered at (4.166,
-23.345, 14.697) was used for the Spike glycoprotein.
Grid box with center at (-13.309, 14.415, 61.16), and
size (30, 40, 40) was used for the main protease. For
ACE2 the grid box we used was with size (126, 88,
68), and centered at (68.719, 73.5, 26.656).
Figure 5: Grid box with AutoDock tools.
5.3 Running AutoDock Vina
As a result from the experiments AutoDock Vina, pro-
duces log file. This file consists of all the poses gener-
ated by the AutoDock Vina along with their binding
affinities and RMSD scores. The first pose is con-
sidered to be the best pose, since it has more binding
affinity than the other poses and is without any RMSD
value. The structure of the file is presented in Fig. 6.
Finding Potential Inhibitors of COVID-19
113

Figure 6: Example of log file that is result from AutoDock
Vina.
6 RESULTS
Tables 1, 2, and 3 present the results obtained
from the experiments. The molecular docking re-
sults showed best dock score with Auranetin with
all three proteins. It has shown affinity of -11.3
with Spike protein, -12.5 with ACE2 and -9.1 affin-
ity with main protease. This is a good indication
to propose Auranetin for further investigation in de-
veloping treatments against COVID-19. Auranetin
belongs to the class of organic compounds known
as 8-o-methylated flavonoids. These are flavonoids
with methoxy groups attached to the C8 atom of the
flavonoid backbone. Thus, Auranetin is considered to
be a flavonoid lipid molecule. Auranetin is a very hy-
drophobic molecule, relatively neutral and practically
insoluble in water. Outside of the human body, au-
ranetin has been detected, but not quantified in, citrus
(Yannai, 2003).
Obacunone, Pomiferin, Eucalyptin and Hes-
peretin showed lower binding energy to Spike protein
active site compared to Remdesivir and Dexametha-
sone. Obacunone is a natural compound present in
citrus fruits. It has been demonstrated for various
biological activities including anti-cancer and anti-
inflammatory properties (Xiang et al., 2015) (Jing
et al., 2019).
Pomiferin can be found along with osajin in the
fruits and female flowers of the osage orange tree
(Maclura pomifera). It is a prenylated isoflavone
that has demonstrated efficacy as an antioxidant, car-
dioprotectant, antimicrobial, antidiabetic, PDE5 in-
hibitor and cytotoxicity for several cancer cell lines
(Gruber et al., 2014).
Eucalyptin has antioxidant and antimicrobial
properties, and also is natural compound. Hesperetin
is also flavonoid, same as Auranetin. This compound,
in the form of its glycoside hesperidin, is the predom-
inant flavonoid in lemons and oranges. It has antiox-
idative, antiinflammatory, and neuroprotective effects
(Hwang et al., 2015).
Most of the compounds that showed highest affin-
ity with these three proteins are natural compounds
that display a large variety of biological activities.
Table 1: Results of molecular docking with Spike protein.
Spike protein
Ligand Affinity (kcal/mol)
Auranetin -11.3
Obacunone -8.1
Pomiferin -7.7
Eucalyptin -7.5
Hesperetin -7.5
Dexamethazone -7.5
Alnetin -7.2
Pedalitin -7.2
Remdesivir -7.2
Citromycetin -7.0
Trifluoperazine -6.8
Tangeretin -6.6
Nobiletin -6.6
Sinensetin -6.5
Beta D Mannose -5.4
Dimetylsulphoxide -2.7
Table 2: Results of molecular docking with ACE2.
ACE2
Ligand Affinity (kcal/mol)
Auranetin -12.5
Obacunone -10.0
Pomiferin -8.9
Dexamethasone -8.3
Pedalitin -8.2
Hesperetin -8.0
Trifluoperazine -7.8
Eucalyptin -7.8
Sinensetin -7.8
Tangeretin -7.7
Remdesivir -7.7
Alnetin -7.6
Nobiletin -7.2
Citromycetin -6.8
Beta D Mannose -5.9
Dimetylsulphoxide -2.4
BIOINFORMATICS 2021 - 12th International Conference on Bioinformatics Models, Methods and Algorithms
114

6.1 Analysing AutoDock Vina Results
with PyMOL
After getting the results from AutoDock Vina, they
can be analysed by using the PyMOL software (Py-
MOL, 2020). It allows the analysis of all the poses
generated from our docking study, starting from the
pose with highest affinity, to the one with lowest affin-
ity (Yuan et al., 2017).
Inspecting the ligand sites, we can see the bonds
that exist between the ligand and the residues of the
protein. If the residue that is part of the interaction is
one of the binding sites of the protein, it means that
the ligand is situated well in the protein’s cavity.
Table 3: Results of molecular docking with Main protease.
Main Protease
Ligand Affinity (kcal/mol)
Auranetin -9.1
Pomiferin -7.2
Obacunone -7.1
Dexamethazone -6.8
Remdesivir -6.3
Pedalitin -6.2
Trifluoperazine -6.2
Hesperetin -6.1
Sinensetin -6.0
Alnetin -5.9
Citromycetin -5.7
Eucalyptin -5.7
Nobiletin -5.7
Tangeretin -5.5
Beta D Mannose -5.2
Dimethylsulphoxide -2.4
Auranetin forms bond with Trp423 amino acid of
the spike glycoprotein. Unfortunately, it is not part of
the active site of the protein. It may be allosteric site,
but this needs to be confirmed by additional experi-
ments and analysis. In biochemistry, regulation of an
enzyme by binding an effector molecule at a site other
than active site, is called allosteric regulation (Coop-
erman, 2013). Effectors that decrease the protein’s
activity are called allosteric inhibitors. The interac-
tion of Auranetin and spike glycoprotein is shown in
Fig. 7.
The bonds formed between the main protease and
auranetin are with the three residues: Lys137, Val202
and Glu288, that again are not part of the active site.
This is shown in Fig. 8. Auranetin forms bond with
Tyr158 amino acid of ACE2, and this can be seen on
Fig. 9.
Analysis of obacunone, the second compound that
Figure 7: Complex of Spike protein and Auranetin with se-
lected ligand sites in PyMOL.
Figure 8: Complex of main protease and Auranetin with
selected ligand sites in PyMOL.
Figure 9: Complex of ACE2 and Auranetin with selected
ligand sites in PyMOL.
showed highest affinity after auranetin, was also done.
It formed two bonds with active site of spike gly-
coprotein, more precisely with Arg495 and Gln401
amino acids. This is shown in Fig. 10. Except au-
ranetin, we suggest doing additional experiments of
obacunone. This compound may also be considered
in developing suitable drug for Sars-CoV-2.
7 CONCLUSIONS
In summary, we have performed the molecular dock-
ing studies of ligands, mostly natural components,
chosen randomly with three important proteins (main
protease, ACE2, and spike glycoprotein) and com-
pared the dock score results with remdesivir and dex-
amethasone. Our results revealed that many of the
Finding Potential Inhibitors of COVID-19
115

Figure 10: Complex of Spike protein with Obacunone with
selected ligand sites in PyMOL.
tested ligands showed higher dock score than remde-
sivir and dexamethasone, with the maximum dock
score shown by auranetin. Therefore, with the com-
bined docking results and the medical importance
of auranetin, we propose that auranetin and other
flavonoids can be further studied with the aim to get
suitable drugs against COVID-19.
REFERENCES
Aaftaab, S., Khusbhoo, J., Sasikala, K., and Mallika, A.
(2019). Molecular docking in modern drug discovery:
Principles and recent applications.
Cooperman, B. S. (2013). Allosteric regulation. Encyclo-
pedia Of Biological Chemistry, 71-74.
FDA (2020). Food and Drug Administration Agency.
Gheblawi, M., Wang, K., Viveiros, A., Nguyen, Q., Zhong,
J.-C., Turner, A. J., et al. (2020). Angiotensin-
converting enzyme 2: Sars-cov-2 receptor and regu-
lator of the renin-angiotensin system. Circ Res. 2020
May 8; 126(10).
Gruber, J. V., Holtz, R., Sikkink, S. K., and Tobin, D. J.
(2014). In vitro and ex vivo examination of topical
pomiferin treatments.
Huang, Y., Yang, C., Xu, X.-f., Xu, W., and Liu, S.-
w. (2020). Structural and functional properties of
sars-cov-2 spike protein: potential antivirus drug de-
velopment for covid-19. Acta Pharmacol Sin 41,
1141–1149 (2020).
Hung, C. and Chen, C. (2014). Computational approaches
for drug discovery. Drug development research, 75(6),
412–418.
Hwang, S.-L., Shih, P.-H., and Yen, G.-C. (2015). Chap-
ter 80 - citrus flavonoids and effects in dementia and
age-related cognitive decline. Diet and Nutrition in
Dementia and Cognitive Decline.
Jean, S.-S., Lee, P.-I., and Hsueh, P.-R. (2020). Treat-
ment options for covid-19: The reality and challenges.
Journal of Microbiology, Immunology and Infection
Volume 53, Issue 3, June 2020, Pages 436-443.
Jing, X., Ai-hua, Z., Shi, Q., Tian-lei, Z., Xian-na, L.,
Guang-li, Y., et al. (2019). Identification of the per-
turbed metabolic pathways associating with prostate
cancer cells and anticancer affects of obacunone.
Kouznetsova, V., Huang, D., and Tsigelny, I. F. (2020).
Potential covid-19 protease inhibitors: Repurposing
fdaapproved drugs.
MGLTools (2020). MGLTools - software developed at
the Molecular Graphics Laboratory (MGL) of The
Scripps Research Institute.
Narkhede, R. R., Cheke, R. S., Ambhore, J. P., and Shinde,
S. D. (2020). The molecular docking study of poten-
tial drug candidates showing anti-covid-19 activity by
exploring of therapeutic targets of sars-cov-2. screen-
ing, 5(8).
Nataraj, S., P., Khajamohiddin, S., and Jack, T. (2017).
Software for molecular docking: a review.
Oleg, T. and Arthur, J. O. (2010). Autodock vina: im-
proving the speed and accuracy of docking with a
new scoring function, efficient optimization and mul-
tithreading. Journal of computational chemistry vol.
31,2 (2010): 455-61.
Omar, S., Bouziane, I., Bouslama, Z., and Djemel, A.
(2020). In-silico identification of potent inhibitors
of covid-19 main protease (mpro) and angiotensin
converting enzyme 2 (ace2) from natural products:
Quercetin, hispidulin, and cirsimaritin exhibited better
potential inhibition than hydroxy-chloroquine against
covid-19 main protease active site and ace2.
Peter, H., Wei, S. L., Jonathan, E., Marion, M., Jennifer,
B., Louise, L., et al. (2020). Effect of dexamethasone
in hospitalized patients with covid-19: Preliminary re-
port. medRxiv.
Pham, M. Q., Vu, K. B., Pham, T. N. H., Tran, L. H., Tung,
N. T., Vu, V. V., Nguyen, T. H., Ngo, S. T., et al.
(2020). Rapid prediction of possible inhibitors for
sars-cov-2 main protease using docking and fpl sim-
ulations. RSC Advances, 10(53):31991–31996.
PubChem (2020). PubChem - open chemistry database at
the National Institutes of Health (NIH).
PyMOL (2020). PyMOL is a user-sponsored molecular
visualization system on an open-source foundation,
maintained and distributed by Schr
¨
odinger.
RCSB (2020). RCSB Protein Data Bank.
Singh, P., Sharma, A., and Nandi, S. P. (2020). Iden-
tification of potent inhibitors of covid-19 main pro-
tease enzyme by molecular docking study. ChemRxiv.
Preprint.
Sparks, D. (2020). Covid-19 (coronavirus) drugs: Are there
any that work? Mayo Clinic.
Ullrich, S. and Nitsche, C. (2020). The sars-cov-2 main pro-
tease as drug target. Bioorganic & Medicinal Chem-
istry Letters, Volume 30, Issue 17, 1 September 2020,
127377.
Vardhan, S. and Sahoo, S. K. (2020). Searching in-
hibitors for three important proteins of covid-19
through molecular docking studies. arXiv preprint
arXiv:2004.08095.
Wu, C., Liu, Y., Yang, Y., Zhang, P., Zhong, W., et al.
(2020). Analysis of therapeutic targets for sars-cov-
BIOINFORMATICS 2021 - 12th International Conference on Bioinformatics Models, Methods and Algorithms
116

2 and discovery of potential drugs by computational
methods. Acta Pharmaceutica Sinica B Volume 10,
Issue 5, May 2020, Pages 766-788.
Xiang, Y., Guodong, D., Xiaoyan, Z., and Hui, X. (2015).
Insight into reduction of obacunone, and their ester
derivatives as insecticidal agents against mythimna
separata walker.
Yamin, M. (2020). Counting the cost of covid-19. Interna-
tional journal of information technology: an official
journal of Bharati Vidyapeeth’s Institute of Computer
Applications and Management, Advance online publi-
cation.
Yannai, S. (2003). Dictionary of food compounds with cd-
rom. new york: Chapman and hall/crc.
Yu, R., Chen, L., Lan, R., Shen, R., and Li, P. (2020). Com-
putational screening of antagonist against the sars-
cov-2 (covid-19) coronavirus by molecular docking.
International Journal of Antimicrobial Agents, page
106012.
Yuan, S., Chan, H. S., and Hu, Z. (2017). Using pymol as a
platform for computational drug design.
Finding Potential Inhibitors of COVID-19
117
