
Lossy Compressor Preserving Variant Calling through Extended BWT
Veronica Guerrini
1 a
, Felipe A. Louza
2 b
and Giovanna Rosone
1 c
1
Department of Computer Science, University of Pisa, Italy
2
Faculty of Electrical Engineering, Federal University of Uberl
ˆ
andia, Brazil
Keywords:
eBWT, LCP, Positional Clustering, FASTQ, Smoothing, Noise Reduction, Compression.
Abstract:
A standard format used for storing the output of high-throughput sequencing experiments is the FASTQ for-
mat. It comprises three main components: (i) headers, (ii) bases (nucleotide sequences), and (iii) quality
scores. FASTQ files are widely used for variant calling, where sequencing data are mapped into a reference
genome to discover variants that may be used for further analysis. There are many specialized compressors
that exploit redundancy in FASTQ data with the focus only on either the bases or the quality scores compo-
nents. In this paper we consider the novel problem of lossy compressing, in a reference-free way, FASTQ data
by modifying both components at the same time, while preserving the important information of the original
FASTQ. We introduce a general strategy, based on the Extended Burrows-Wheeler Transform (EBWT) and
positional clustering, and we present implementations in both internal memory and external memory. Exper-
imental results show that the lossy compression performed by our tool is able to achieve good compression
while preserving information relating to variant calling more than the competitors.
Availability: the software is freely available at https://github.com/veronicaguerrini/BFQzip.
1 INTRODUCTION
The recent improvements in high-throughput se-
quencing technologies have led a reduced cost of
DNA sequencing and unprecedented amounts of ge-
nomic datasets, which has motivated the development
of new strategies and tools for compressing these data
that achieve better results than general-purpose com-
pression tools – see (Numanagi
´
c et al., 2016; Hernaez
et al., 2019) for good reviews.
FASTQ is the standard text-based format used to
store raw sequencing data, each DNA fragment (read)
is stored in a record composed by three main com-
ponents: (i) read identifier with information related
to the sequencing process (header), (ii) nucleotide
sequence (bases), and (iii) quality sequence, with a
per-base estimation of sequencing confidence (qual-
ity scores). The last two components are divided by
a “separator” line, which is generally discarded by
compressors as it contains only a “+” symbol option-
ally followed by the same header.
The majority of compressors for FASTQ files
commonly split the data into those three main com-
a
https://orcid.org/0000-0001-8888-9243
b
https://orcid.org/0000-0003-2931-1470
c
https://orcid.org/0000-0001-5075-1214
ponents (or streams), and compress them separately,
which allows much better compression rates.
The headers can be efficiently compressed taking
advantage of their structure and high redundancy. A
common strategy used by FASTQ compressors, like
SPRING (Chandak et al., 2018) and FaStore (Roguski
et al., 2018), is to tokenize each header: the sepa-
rators are non-alphanumerical symbols (Bonfield and
Mahoney, 2013).
The bases and quality scores are commonly pro-
cessed separately, although their information are cor-
related, and current specialized compressors only fo-
cus on one of these two components.
Most of FASTQ compressors limit their focus on
the bases, compressing the quality scores indepen-
dently with a third tool or standard straightforward
techniques. The approaches that focus on compres-
sion the bases component are lossless, i.e., they do
not modify the bases, but they find a good strategy to
represent the data by exploiting the redundancy of the
DNA sequences. An interesting strategy is to reorder
the sequences in the FASTQ file to gather reads orig-
inating from close regions of the genome (Cox et al.,
2012; Roguski et al., 2018; Hach et al., 2012; Chan-
dak et al., 2018).
38
Guerrini, V., Louza, F. and Rosone, G.
Lossy Compressor Preserving Variant Calling through Extended BWT.
DOI: 10.5220/0010834100003123
In Proceedings of the 15th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2022) - Volume 3: BIOINFORMATICS, pages 38-48
ISBN: 978-989-758-552-4; ISSN: 2184-4305
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

On the other hand, approaches that focus on com-
pression the quality score component are generally
lossy, i.e., they modify the data by smoothing the
quality scores whenever possible. These approaches
can be reference-based when they use external infor-
mation (besides the FASTQ itself), such as a reference
corpus of k-mers, e.g. QUARTZ (Yu et al., 2015),
GeneCodeq (Greenfield et al., 2016) and YALFF
(Shibuya and Comin, 2019). While, reference-free
strategies evaluate only the quality scores informa-
tion, such as the quantization of quality values using
QVZ (Malysa et al., 2015), Illumina 8-level binning
and binary thresholding, or evaluate the related bio-
logical information in the bases component (strate-
gies known as read-based) – e.g. BEETL (Janin et al.,
2014) and LEON (Benoit et al., 2015).
Our Contribution. In this work, we focus on a
novel approach for the lossy compression of both the
bases and the quality scores components taking into
account both information at the same time. In fact,
the two components are highly correlated being the
second one a confidence estimation of each base call
contained in the first one. To the best of our knowl-
edge, none of the existing FASTQ compressor tools
evaluates and modifies both components at the same
time.
Note that we are not interested in compressing the
headers component, for which one can use any state-
of-the-art strategies or ignore them. Indeed, headers
can be artificially structured in fields to store informa-
tion only related to the sequencing process.
We focus on lossy reference-free and read-based
FASTQ compression that makes clever modifications
on the data, by reducing noise in the bases compo-
nent that could be introduced by the sequencer, and
by smoothing irrelevant values on the quality scores
component, according to the correlated information
in the bases, that would guarantee to preserve variant
calling.
Hence, we propose a novel read-based, reference-
and assembly-free compression approach for FASTQ
files, BFQZIP, which combines both DNA bases and
quality information for obtaining a lossy compressor.
Similarly to BEETL (Janin et al., 2014), our ap-
proach is based on the Extended Burrows-Wheeler
Transform (EBWT) (Burrows and Wheeler, 1994;
Mantaci et al., 2007) and its combinatorial properties,
and it applies the idea that each base in a read can with
high probability be predicted by the context of bases
that are next to it. We also exploit the fact that such
predicted bases add little information, and its quality
score can be discarded or heavily compressed without
distortion effects on downstream analysis.
In our strategy, the length of contexts is variable-
order (i.e. not fixed a priori, unlike BEETL), and can
be as large as the full read length for high-enough cov-
erages and small-enough error rates.
We exploit the positional clustering framework in-
troduced in (Prezza et al., 2019) to detect “relevant”
blocks in the EBWT. These blocks allow us not only
to smooth the quality scores, but also to apply a noise
reduction on the corresponding bases, replacing those
that are believed to be noise, while keeping variant
calling performance comparable to that with the orig-
inal data and with compression rates comparable to
other tools.
2 PRELIMINARIES
Let S be a string (also called sequence or reads due to
our target application) of length n on the alphabet Σ.
We denote the i-th symbol of S by S[i]. A substring
of any S ∈ S is denoted as S[i, j] = S[i]···S[ j], with
S[1, j] being called a prefix and S[i,n + 1] a suffix of S.
Let S = {S
1
,S
2
,. . . , S
m
} be a collection of m
strings. We assume that each string S
i
∈ S has length
n
i
and is followed by a special end-marker symbol
S
i
[n
i
+1] = $
i
, which is lexicographically smaller than
any other symbol in S , and does not appear in S else-
where.
The Burrows-Wheeler Transform (BWT) (Bur-
rows and Wheeler, 1994) of a text T (and the EBWT
of a set of strings S (Mantaci et al., 2007; Bauer
et al., 2013)) is a suitable permutation of the sym-
bols of T (and S ) whose output shows a local simi-
larity, i.e. symbols preceding similar contexts tend to
occur in clusters. Both transformations have been in-
tensively studied from a theoretical and combinatorial
viewpoint and have important and successful applica-
tions in several areas, e.g. (Mantaci et al., 2008; Li
and Durbin, 2010; Kimura and Koike, 2015; Shibuya
and Comin, 2019; Gagie et al., 2020; Guerrini et al.,
2020).
We assume that N =
∑
m
i=1
(n
i
+1) denotes the sum
of the lengths of all strings in S . The output of
the EBWT is a string ebwt(S ) of length N such that
ebwt(S )[i] = x, with 1 ≤ i ≤ N, if x circularly pre-
cedes the i-th suffix (context) S
j
[k, n
j
+ 1] (for some
1 ≤ j ≤ m and 1 ≤ k ≤ n
j
+1), according to the lexi-
cographic sorting of the contexts of all strings in S . In
this case, we say that the context S
j
[k, n
j
+ 1] is asso-
ciated with the position i in ebwt(S ). See Table 1 for
an example.
In practice, computing the EBWT via suffix sort-
ing (Bauer et al., 2013; Bonomo et al., 2014) may be
done considering the same end-markers for all strings.
Lossy Compressor Preserving Variant Calling through Extended BWT
39

Table 1: Extended Burrows-Wheeler Transform (EBWT), LCP array, and auxiliary data structures used for detecting posi-
tional clusters for the set S = {GGCGTACCA$
1
,GGGGCGTAT $
2
,ACGANTACGAC$
3
} and k
m
= 2.
i B
min
B
thr
lcp ebwt Sorted Suffixes i B
min
B
thr
lcp ebwt Sorted Suffixes
1 0 0 0 A $
1
17 0 1 4 G C G T A T $
2
2 0 0 0 T $
2
18 1 0 0 C G A C$
3
3 0 0 0 C $
3
19 0 1 2 C G A N T A C G A C $
3
4 0 0 0 C A$
1
20 1 0 1 G G C G T A C C A$
1
5 0 0 1 G A C$
3
21 0 1 5 G G C G T A T $
2
6 0 1 2 T A C C A$
1
22 1 0 1 $ GG C G T A C C A$
1
7 0 1 2 T A C G A C $
3
23 0 1 6 G G G C G T A T $
2
8 0 1 4 $ A C G A N T A C GAC $
3
24 1 1 2 G G G G C G T A T $
2
9 1 0 1 G A N T A C GA C $
3
25 0 1 3 $ GGGG C G T A T $
2
10 0 0 1 T A T $
2
26 1 0 1 C G T A C C A$
1
11 1 0 0 A C$
3
27 0 1 3 C G T A T $
2
12 0 0 1 C C A$
1
28 1 0 0 A N T A C G A C $
3
13 0 0 1 A C C A$
1
29 0 0 0 A T$
2
14 0 0 1 A C G A C $
3
30 0 0 1 G T A C C A$
1
15 0 1 3 A C G A N T A C G AC$
3
31 0 1 3 N T A C G A C $
3
16 1 1 2 G C G T A C C A$
1
32 0 1 2 G T A T$
2
That is, we assume that $
i
< $
j
, if i < j, and use a
unique symbol $ as the end-marker for all strings.
The longest common prefix (LCP) array (Manber
and Myers, 1990) of S is the array lcp(S ) of length
N +1, such that for 2 ≤ i ≤ N, lcp(S )[i] is the length of
the longest common prefix between the contexts asso-
ciated with the positions i and i − 1 in ebwt(S ), while
lcp(S )[1] = lcp(S )[N + 1] = 0. The LCP-intervals
(Abouelhoda et al., 2004) are maximal intervals [i, j]
that satisfy lcp(S )[r] ≥ k for i < r ≤ j and whose as-
sociated contexts share at least the first k symbols.
An important property of the BWT and EBWT,
is the so-called LF mapping (Ferragina and Manzini,
2000), which states that the i-th occurrence of sym-
bol x on the BWT string and the first symbol of the
i-th lexicographically-smallest suffix that starts with
x correspond to the same position in the input string
(or string collection). We will use the LF mapping
to perform backward searches when creating a new
(modified) FASTQ file. The backward search allows
to find the range of suffixes prefixed by a given string
– see (Ferragina and Manzini, 2000; Adjeroh et al.,
2008) for more details.
3 METHOD
We structure our reference-free FASTQ compression
method in four main steps: (a) data structures build-
ing, (b) positional cluster detecting, (c) noise reduc-
tion and quality score smoothing, and (d) FASTQ re-
construction.
(a) Data Structures Building. This phase consists
in computing the EBWT and the LCP array for the
collection of sequences S stored in the bases compo-
nent of the input FASTQ file. We also compute qs(S ),
as the concatenation of the quality scores associated
with each symbol in ebwt(S ), i.e., the string qs con-
tains a permutation of the quality score symbols that
follows the symbol permutation in ebwt(S ). Note that
the lcp(S ) is only used in the next step, so we can ei-
ther explicitly compute it in this phase or implicitly
deduce it by ebwt(S ) during the next step.
(B) Positional Cluster Detecting. A crucial prop-
erty of the EBWT is that symbols preceding suffixes
that begin with the same substring (context) w will
result in a contiguous substring of ebwt, and thus of
qs. Such a substring of the ebwt is generally called
cluster. In literature, such clusters depending on the
length k of the context w are associated with LCP-
intervals (Abouelhoda et al., 2004).
The aim of the positional clustering frame-
work (Prezza et al., 2019; Prezza et al., 2020) is to
overcome the limitation of strategies based on LCP-
intervals, which depend on the choice of k. Intuitively,
meaningful clusters in the EBWT lie between local
minima in the LCP array, and symbols of the same
positional cluster usually cover the same genome lo-
cation (Prezza et al., 2019). This recent strategy au-
tomatically detects, in a data-driven way, the length k
of the common prefix shared by the suffixes of a clus-
ter in the EBWT. Moreover, so as to exclude clusters
corresponding to short random contexts, we set a min-
imum length for the context w, denoted by k
m
.
Analogously to (Prezza et al., 2020), we define
positional clusters by using two binary vectors: B
thr
and B
min
, where B
thr
[i] = 1 if and only if lcp[i] ≥ k
m
,
and B
min
= 1 if and only if lcp[i] is a local mini-
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
40

mum i.e., it holds lcp[i − 1] > lcp[i] ≤ lcp[i +1], for all
1 < i ≤ N, which depends on data only. A EBWT po-
sitional cluster is a maximal substring ebwt[i, j] such
that B
thr
[r] = 1, for all i < r ≤ j, and B
min
[r] = 0, for
all i < r ≤ j. See, for instance, Table 1.
(C.1) Noise Reduction. Given the base symbols ap-
pearing in any positional cluster of ebwt(S ), we call
as frequent symbol any symbol whose occurrence in
the cluster is greater than a threshold percentage. The
idea that lies behind changing bases is to reduce the
number of symbols in a cluster that are different from
the most frequent symbols, while preserving the vari-
ant calls. So, we take into account only clusters that
have no more than two frequent symbols (for exam-
ple, we set the threshold percentage to 40%).
The symbols in an EBWT positional cluster usu-
ally correspond to the same genome location (Prezza
et al., 2019). Thus, given an EBWT positional cluster
α = ebwt[i, j], we say a symbol b is a noisy base if
it is different from the most frequent symbols and all
occurrences of b in α are associated with low qual-
ity values in qs[i, j] (i.e. there are no occurrences of
b with a high quality score in α). Intuitively, a noisy
base is more likely noise introduced during the se-
quencing process.
Then, for each analyzed cluster α, we replace
noisy bases in α with a predicted base c as follows.
We distinguish two cases. If the cluster α contains a
unique most frequent symbol c, then we replace the
noisy base b with c. Otherwise, if we have two dif-
ferent frequent symbols, for each occurrence of them
and for the noisy base b, we compute the preceding
context of length ` in their corresponding reads (i.e.
left context of each considered base), by means of the
backward search applied to ebwt(S) (for example, we
set ` = 1 in our experiments). If the left context pre-
ceding b coincides with the contexts preceding all the
occurrences of d (one of the two most frequent sym-
bols), then we replace the base b with d. We spec-
ify that no base changes are performed if the frequent
symbols are preceded by the same contexts.
(C.2) Quality Score Smoothing. During step (c.1),
we also modify quality scores by smoothing the sym-
bols of qs that are associated with base symbols in
clusters of ebwt.
In any cluster α, the value Q used for replacements
can be computed with different strategies: (i) Q is a
default value, or (ii) Q is the quality score associated
with the mean probability error in α, or (iii) Q is the
maximum quality score in α, or (iv) Q is the average
of the quality scores in α. According to this smooth-
ing process, apart from strategy (i), the value Q de-
pends on the cluster analyzed. In the experiments we
evaluated qs smoothing as follows. For each position
r within α = ebwt[i, j] we smooth the quality score
qs[r] with Q, either if ebwt[r] is one of the most fre-
quent symbols (regardless of its quality), or if qs[r] is
greater than Q.
An additional feature to compress further quality
scores is the possibility of reducing the number of the
alphabet symbols appearing in qs(S ). This smooth-
ing approach is quite popular and standard in litera-
ture (Chandak et al., 2018). Then, in addition to one
of the strategies described above, we can apply the Il-
lumina 8-level binning reducing to 8 the number of
different symbols in qs.
(D) FASTQ Reconstruction and Compression.
Given the modified symbols in ebwt(S ) and in qs(S )
(according to the strategies described above), we use
the LF mapping on the original ebwt string to retrieve
the order of symbols and output a new (modified)
FASTQ file.
The headers component with the read titles can be
either omitted (inserting the symbol ‘@’ as header) or
kept as they are in the original FASTQ file.
At the end, the resulting FASTQ file is com-
pressed by using any state-of-the-art compressor.
4 EXPERIMENTS
Implementations. We present two implementa-
tions of our tool BFQZIP: in internal memory and
in external memory. Now we give a brief description
of them.
Given a FASTQ file containing a collection S ,
both implementations take as input the files contain-
ing ebwt(S ) and qs(S ). In the external memory, we
also need the array lcp(S ).
The construction of these data structures dur-
ing the first step can be performed with any tools.
e.g. (Bauer et al., 2013; Bonizzoni et al., 2019; Egidi
et al., 2019; Louza et al., 2020; Boucher et al., 2021)
according to the resources available (which is a good
feature).
The two implementations largely differ in the de-
tection of positional clusters. Indeed, alike (Prezza
et al., 2020), the internal memory approach rep-
resents ebwt(S ) via the compressed suffix tree de-
scribed in (Prezza and Rosone, 2021) (see also (Be-
lazzougui et al., 2020)), where it is shown that lcp(S )
can be induced from the EBWT using succinct work-
ing space for any alphabet size. Whereas, in exter-
nal memory, EBWT positional clusters are detected
Lossy Compressor Preserving Variant Calling through Extended BWT
41

Table 2: Paired-end datasets used in the experiments and their sizes in bytes. Each dataset is obtained from two files ( 1 and
2), whose number of reads and read length are given in columns 2 and 3. We distinguish the size of the original FASTQ (raw
data) from the size of the same FASTQ file with all headers removed (i.e., replaced by ’@’). In the last column we report the
size of the bases component, that is equal to the quality scores component size.
Dataset No. reads Length Raw (complete) FASTQ DNA/QS
ERR262997 1 - chr 20 13,796,697 101 3,420,752,544 2,869,712,976 1,407,263,094
ERR262997 2 - chr 20 13,796,697 101 3,420,752,544 2,869,712,976 1,407,263,094
ERR262997 1 - chr 14 18,596,541 101 4,611,888,574 3,868,080,528 1,896,847,182
ERR262997 2 - chr 14 18,596,541 101 4,611,888,574 3,868,080,528 1,896,847,182
ERR262997 1 - chr 1 49,658,795 101 12,211,743,094 10,329,029,360 5,065,197,090
ERR262997 2 - chr 1 49,658,795 101 12,211,743,094 10,329,029,360 5,065,197,090
by reading the lcp(S ) stored in a file in a sequential
way.
For the last two steps, the two implementations
are similar, except that the data are kept in internal or
external memory. In particular, during step (d), we
use the LF-mapping either in internal memory – via
the suffix-tree navigation as in (Prezza et al., 2020)
– or in external memory – similarly to (Bauer et al.,
2013).
Datasets. The easiest approach to evaluate the va-
lidity of our method could be to simulate reads and
variants from a reference genome. However, it is not
trivial to simulate variant artifacts for this purpose (Li,
2014), so we focus only on real data.
In this study, thus, we use the real human dataset
ERR262997 corresponding to a 30x-coverage paired-
end Whole Genome Sequencing (WGS) data for the
CEPH 1463 family. Similarly to other studies (Ochoa
et al., 2016), for evaluation purposes we extracted the
chromosomes 20, 14 and 1 from ERR262997, obtain-
ing datasets of different sizes (see Table 2).
Compression. We describe experiments that show
that our strategy is able to compress FASTQ files in
lossy way modifying both the bases and quality scores
component, while keeping most of the information
contained in the original file.
To the best of our knowledge, none of the existing
FASTQ compressors evaluates and modifies both the
bases and the quality scores components at the same
time. Therefore, no comparison with existing tools is
completely fair. We consider the tools BEETL
1
(Janin
et al., 2014) and LEON
2
(Benoit et al., 2015) that are
reference-free and read-based: they smooth the qual-
ity score component in a lossy way based on the bi-
ological information of the bases, without modifying
the bases themselves.
1
https://github.com/BEETL/BEETL/blob/
RELEASE 1 1 0/scripts/lcp/applyLcpCutoff.pl
2
http://gatb.inria.fr/software/leon/
We choose BEETL, because it is based on EBWT
and takes as input the same data structures we com-
pute during step (a). BEETL smooths to a con-
stant value the quality scores corresponding to each
run of the same symbol associated with the LCP-
interval [i, j]. The quality scores associated to each
run in ebwt[i, j] are smoothed if the length of the run is
greater than a minimum stretch length s. So, it needs
two parameters: the minimum stretch length s and the
cut threshold c for the LCP-interval, failing to sepa-
rate read suffixes differing after c positions (this is,
indeed, a drawback of all c-mer-based strategies).
We choose LEON (Benoit et al., 2015) that, on
the contrary, needs to build a reference from the in-
put reads in the form of a bloom filter compressed
de Bruijn graph, and then maps each nucleotide se-
quence as a path in the graph. Thus, LEON can be
considered assembly-based, since it uses a de Bruijn
graph as a de novo reference. If a base is covered by
a sufficiently large number of k-mers (substrings of
length k) stored in the bloom filter, its quality is set to
a fixed high value. Thus, LEON depends on a fixed
parameter k for the graph, as well.
Regarding the output of each tool, BFQZIP pro-
duces a new FASTQ file with modified bases and
smoothed qualities, whereas BEETL only produces a
BWT-ordered smoothed qualities file, which is used
to replace the quality scores component in the modi-
fied FASTQ file (with the original bases component).
LEON produces a proprietary format compressed file
that encodes the de Bruijn graph, thus, it was neces-
sary to uncompress the output file to obtain the mod-
ified FASTQ. For a fairer comparison, these resulting
FASTQ files were compressed with the same tools.
In particular, we choose two well-known compressors
for this task: PPMd (Cleary and Witten, 1984; Moffat,
1990) and BSC
3
.
We run BFQZIP and BEETL with similar param-
eters: in BEETL, we set the replacement quality score
to ‘@’ (as set by LEON) and the minimum LCP cut
threshold to 30. For BFQZIP, we used options -T 30
to set the minimum context length k
m
= 30 and -Q
3
http://libbsc.com/
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
42

Table 3: Compression ratio for (original and three smoothing tools) FASTQ files (with headers replaced by ’@’) and their
single components (qualities (QS) and bases (DNA)) obtained by both PPMd and BSC. The ratio is defined as compressed
size/original size, where original file size is in Table 2. Since BEETL and LEON do not modify the bases component, their
ratio for the DNA component is the same as the original.
Tool
FASTQ QS DNA FASTQ QS DNA
ERR262997 1 chr 20 ERR262997 2 chr 20
PPMd
Original 0.2473 0.2999
0.2037
0.2547 0.3142
0.2046LEON 0.1152 0.0317 0.1234 0.0473
BEETL 0.1900 0.1833 0.2002 0.2033
BFQZIP 0.1941 0.1918 0.2035 0.2043 0.2119 0.2043
BSC
Original 0.2005 0.2902
0.1154
0.2095 0.3034
0.1207LEON 0.0677 0.0241 0.0780 0.0394
BEETL 0.1413 0.1724 0.1534 0.1915
BFQZIP 0.1453 0.1830 0.1152 0.1572 0.2024 0.1200
ERR262997 1 chr 14 ERR262997 2 chr 14
PPMd
Original 0.2482 0.2956
0.2100
0.2544 0.3076
0.2106LEON 0.1175 0.0301 0.1249 0.0444
BEETL 0.1916 0.1805 0.2010 0.1989
BFQZIP 0.1957 0.1889 0.2098 0.2050 0.2074 0.2103
BSC
Original 0.1992 0.2862
0.1174
0.2071 0.2972
0.1224LEON 0.0674 0.0226 0.0770 0.0367
BEETL 0.1406 0.1698 0.1518 0.1874
BFQZIP 0.1445 0.1786 0.1164 0.1555 0.1962 0.1210
ERR262997 1 chr 1 ERR262997 2 chr 1
PPMd
Original 0.2461 0.2969
0.2046
0.2529 0.3097
0.2054LEON 0.1148 0.0299 0.1224 0.0445
BEETL 0.1876 0.1777 0.1973 0.1966
BFQZIP 0.1918 0.1864 0.2044 0.2015 0.2054 0.2051
BSC
Original 0.1984 0.2874
0.1146
0.2068 0.2991
0.1197LEON 0.0661 0.0224 0.0758 0.0367
BEETL 0.1379 0.1669 0.1494 0.1850
BFQZIP 0.1419 0.1759 0.1136 0.1533 0.1941 0.1183
Table 4: Evaluation of called variants by means of rtg vcfeval: comparison between called variants from a modified
FASTQ and variants from the original FASTQ used as baseline.
ERR262997 chr 20 ERR262997 chr 14 ERR262997 chr 1
PREC SEN F PREC SEN F PREC SEN F
LEON 0.9593 0.9356 0.9473 0.9626 0.9381 0.9502 0.9589 0.9348 0.9467
BEETL 0.9584 0.9529 0.9556 0.9626 0.9553 0.9590 0.9596 0.9526 0.9561
BFQZIP 0.9613 0.9534 0.9574 0.9650 0.9555 0.9602 0.9628 0.9523 0.9575
@ to set the constant replacement value. LEON was
executed with default parameters for k-mer size and
minimal abundance threshold, as suggested by the au-
thors. The exact commands for the tools are:
• python3 BFQzip.py <input>.fastq -o
<output>.fastq -T 30 -Q @
• applyLcpCutoff.pl -b <input>.ebwt
-q <input>.ebwt.qs -l <input>.lcp -o
<output>.ebwt.qs -c 30 -r 64 -s 5
• leon -file <input>.fastq -c (for decom-
pression -d) -nb-cores 1
In Table 3, we report the compression ratios
achieved by PPMd and BSC given as input the
FASTQ modified by any of the three tools and the
original FASTQ file. Note that each of the two
FASTQ files comprising any paired-end dataset is
compressed separately.
Table 3 shows that all tools improve the compres-
sion of the data (compared with the original FASTQ).
In particular, LEON is better in terms of compres-
sion ratios than other tools. This improvement is
due to a greater ability to smooth the quality scores
component. Recall that LEON truncates all qual-
ity scores above a given threshold (qualities higher
than ‘@’ are replaced by ‘@’) and in these datasets
the total frequency of symbols greater than ‘@’ is
about 80− 90%. However, the results of BFQZIP and
BEETL were similar in almost all cases.
Validation. In lossy FASTQ compression, it is im-
portant to take into account the impact of the modified
data on downstream analysis, so we need to evaluate
the genotyping accuracy.
Lossy Compressor Preserving Variant Calling through Extended BWT
43

Figure 1: ROC Curves obtained by rtg rocplot: true positive as a function of the false positive respect both to the Ground
truth as baseline (left side) and to the original file as baseline (right side).
We compared the set of variants retrieved from
a baseline with the set retrieved from the modified
FASTQ. First, we considered as baseline the set of
“ground truth” variants for NA12878 provided by Il-
lumina
4
and then, we considered as baseline the set of
variants obtained from the original FASTQ files.
4
https://github.com/Illumina/PlatinumGenomes
The SNP calling pipeline is a bash script
5
(Li, 2013) to align sequences to the reference (in
our case, the latest build of the human reference
genome, GRCh38/hg38) and GATK-HaplotypeCaller
(DePristo and et al., 2011) to call SNPs. The output
5
https://github.com/veronicaguerrini/BFQzip/blob/
main/variant calling/pipeline SNPsCall.sh
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
44
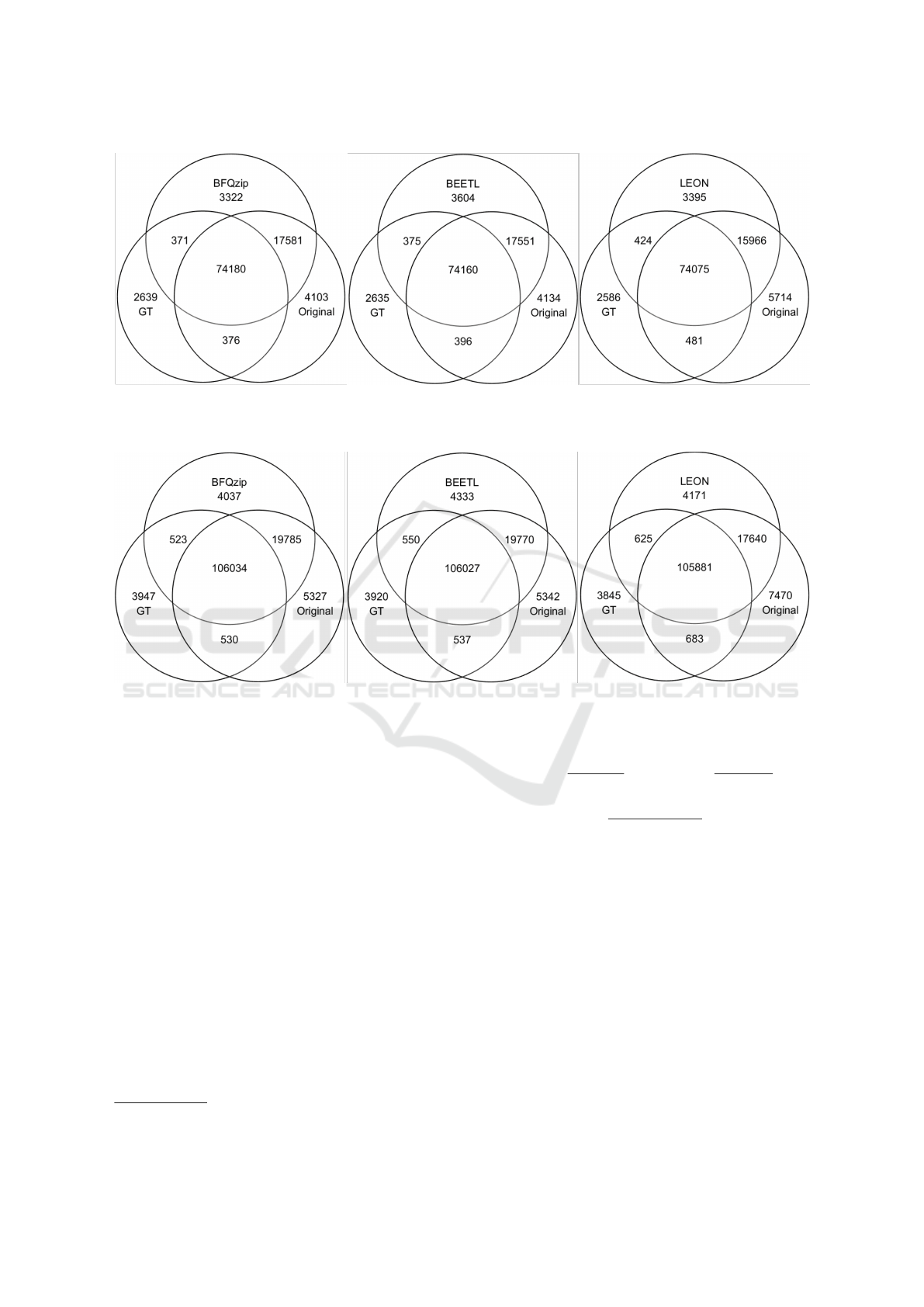
Figure 2: Venn diagrams for chr 20 variants: set comparison between the variants in the original FASTQ file (Original), those
in the ground truth (GT) and those called from the FASTQ modified by BFQzip (left), or modified by BEETL (middle), or
modified by LEON (right).
Figure 3: Venn diagrams for chr 14 variants: set comparison between the variants in the original FASTQ file (Original), those
in the ground truth (GT) and those called from the FASTQ modified by BFQzip (left), or modified by BEETL (middle), or
modified by LEON (right).
of the pipeline is a VCF file that contains the set
of called SNPs, which are then compared to those
contained in a baseline set by using RTG Tools
6
:
rtg vcfeval evaluates agreement between called
and baseline variants using the following performance
metrics.
True positive (T P) are those variants that match
in both baseline and query (the set of called variants);
false positives (FP) are those variants that have mis-
matching, that are in the called set of variants but
not in the baseline; false negatives (FN) are those
variants that are present in the baseline, but miss-
ing in the query. These values are employed to
compute precision (PREC) that measures the propor-
tion of called variants that are true, and sensitivity
(SEN), which measures the proportion of called vari-
ants included in the consensus set. A third metric is
the harmonic mean between sensitivity and precision
(known as F-measure):
6
https://www.realtimegenomics.com/products/rtg-tools
PREC =
T P
T P + FP
, SEN =
T P
T P + FN
,
F =
2 · SEN · PREC
SEN + PREC
.
The tool rtg vcfeval can also output a ROC file
based on the QUAL field, which can be viewed by rtg
rocplot.
Table 4 reports the evaluation results of the vari-
ants retrieved from a modified FASTQ (produced by
any tool) compared to the set of variants from the
original (unsmoothed) FASTQ, which is used as base-
line. We observe that our method provides the highest
precision and F measure. This means that, with re-
spect to the original FASTQ, we have a higher num-
ber of common variants (TP) and the lowest number
of newly-introduced variants (FP), thus we preserve
information of the original FASTQ more than the oth-
ers (see also Figure 1, right side). This is a desirable
property useful in several applications.
Lossy Compressor Preserving Variant Calling through Extended BWT
45

In Figure 1 we provide the ROC curves associ-
ated with the variant comparison performed by rtg
vcfeval (reporting the number of TP as a function of
the FP), showing similar results.
Figure 1 (left side) shows that our tool preserves
those variants that are in the ground truth: using the
VCF file of the ground truth as baseline, all tools
show an accuracy similar to the original. In particular,
BFQZIP and BEETL curves in each graph overlap the
original one, while LEON’s curve is a bit lower.
Figure 1 (right side) also shows that our tool pre-
serves the original variants (TP) introducing only a
little number of new ones (FP) when the original
FASTQ variants are used as baseline (as also shown
by the percentages of sensitivity and precision in Ta-
ble 4) .
With the idea of showing the preservation of vari-
ants from the original file and the possibility of los-
ing variants due to the sequencer noise, we decided to
manually check the variants obtained for any tool by
intersecting the corresponding vcf file with that from
the FASTQ original and with the ground truth vcf.
Figures 2 and 3 show as our tool preserves the vari-
ants that are both in the original file and in the ground
truth (i.e. GT) more than the others.
This analysis appears to confirm what we ob-
served with the ROC curves. BFQZIP reports the
smaller number of new variants introduced by the
tool, that are those variants neither in the original file
nor in the ground truth.
The majority of the variants present in both the
original FASTQ and the ground truth is preserved by
all tools. However, it appears that the heavy trunca-
tion of the quality scores carried out by LEON leads to
a loss of variants present in the original file (see, for
instance, the intersection between each tool and the
original, or the intersection between the ground truth
and the original). While BFQZIP (and in the simi-
lar way BEETL) preserves a high number of variants
that are present in the original file (see intersection
between each tool and the original).
We also made a detailed analysis of the modifica-
tions performed by our method, comparing the modi-
fied bases which correspond to parts aligned by BWA
with the relative bases in the reference (taking into ac-
count how BWA aligned the reads). We have noticed
that about 91-93% of the changed bases correspond to
the reference. The remaining part includes positions
we cannot evaluate, such as those in the reads skipped
by the aligner.
5 DISCUSSION AND
CONCLUSIONS
We propose the first lossy reference-free and
assembly-free compression approach for FASTQ
files, which combines both DNA bases and quality in-
formation in the reads to smooth the quality scores
and to apply a noise reduction of the bases, while
keeping variant calling performance comparable to
that with original data. To the best of our knowledge,
there are no tools that have been designed for this pur-
pose, so we compared our results with two reference-
free and read-based tools that only smooth out the
quality scores component: BEETL (Janin et al., 2014)
and LEON (Benoit et al., 2015).
The resulting FASTQ file with the modified bases
and quality scores produced by our tool achieves bet-
ter compression than the original data. In particular,
by using our approach the bases component achieves
better compression than the original (therefore also
than competitors which do not make any changes to
this component), whereas the compression ratio of the
quality scores component is more competitive with
BEETL than LEON, which also truncates all quality
values greater than ‘@’. On the other hand, in terms
of variant calling, our tool keeps the same accuracy
as the original FASTQ data when the ground truth is
used as baseline, and preserves the variant calls of the
original FASTQ file better than BEETL and LEON.
From the viewpoint of the used resources, LEON
has shown to be the fastest tool, although this com-
parison is not completely fair because our tool mod-
ifies different components of the FASTQ file and the
outputs are different (not directly comparable). More-
over, the authors in (Benoit et al., 2015) state that for
WGS datasets, the relative contribution of the Bloom
filter is low for high coverage datasets, but prohibitive
for low coverage datasets (e.g. 10x). We intend to
improve our implementation also using, for instance,
parallelization, and test our tool for low coverage
datasets and longer reads.
Our implementations give as output the modified
FASTQ file, so we have used PPMd and BSC for
compression, but we could use any other compres-
sor for this task and we could also combine exist-
ing lossless compression schemes to further reduce
the size of the FASTQ file, for instance we could use
SPRING (Chandak et al., 2018), FQSquezeer (De-
orowicz, 2020), and others.
Moreover, our strategy could take advantage of
the reordering of the reads based on their similarity,
e.g. as in (Cox et al., 2012; Chandak et al., 2018;
Deorowicz, 2020). Another feature we did not ex-
ploit in our compression scheme is the paired-end in-
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
46

formation coming from reads in pairs. (Indeed, we
compress the FASTQ files in a paired-end dataset in-
dependently, as they were single-end.) Both the above
aspects could be analyzed as future work.
We believe the results presented in this paper can
motivate the development of new FASTQ compres-
sors that modify the bases and quality scores com-
ponents taking into account both information at the
same time to achieve better compression while keep-
ing most of the relevant information in the FASTQ
data. As future work we intend to investigate the er-
ror correction problem that needs to take into account
much more information (e.g. reverse-complement, or
paired-end information).
ACKNOWLEDGEMENTS
The authors would like to thank N. Prezza for valu-
able comments and suggestions and for providing part
of the code library, and E. Niccoli for preliminary ex-
perimental investigations on positional clustering and
compression in his bachelor’s thesis under the super-
vision of GR and VG.
Work partially supported by the project MIUR-
SIR CMACBioSeq (“Combinatorial Methods for
Analysis and Compression of Biological Sequences”)
grant n. RBSI146R5L and by the University of Pisa
under the “PRA – Progetti di Ricerca di Ateneo” (In-
stitutional Research Grants) - Project no. PRA 2020-
2021 26 “Metodi Informatici Integrati per la Biomed-
ica”.
REFERENCES
Abouelhoda, M. I., Kurtz, S., and Ohlebusch, E. (2004). Re-
placing suffix trees with enhanced suffix arrays. Jour-
nal of Discrete Algorithms, 2(1):53 – 86.
Adjeroh, D., Bell, T., and Mukherjee, A. (2008). The
Burrows-Wheeler Transform: Data Compression,
Suffix Arrays, and Pattern Matching. Springer.
Bauer, M., Cox, A., and Rosone, G. (2013). Lightweight
algorithms for constructing and inverting the BWT of
string collections. Theor. Comput. Sci., 483(0):134 –
148.
Belazzougui, D., Cunial, F., K
¨
arkk
¨
ainen, J., and M
¨
akinen,
V. (2020). Linear-time string indexing and analysis in
small space. ACM Trans. Algorithms, 16(2).
Benoit, G., Lemaitre, C., Lavenier, D., Drezen, E., Dayris,
T., Uricaru, R., and Rizk, G. (2015). Reference-free
compression of high throughput sequencing data with
a probabilistic de bruijn graph. BMC Bioinformatics,
16.
Bonfield, J. K. and Mahoney, M. V. (2013). Compression of
FASTQ and sam format sequencing data. PLOS ONE,
8(3).
Bonizzoni, P., Della Vedova, G., Pirola, Y., Previtali,
M., and Rizzi, R. (2019). Multithread Multistring
Burrows-Wheeler Transform and Longest Common
Prefix Array. Journal of computational biology,
26(9):948—961.
Bonomo, S., Mantaci, S., Restivo, A., Rosone, G., and
Sciortino, M. (2014). Sorting conjugates and suffixes
of words in a multiset. International Journal of Foun-
dations of Computer Science, 25(08):1161–1175.
Boucher, C., Cenzato, D., Lipt
´
ak, Z., Rossi, M., and
Sciortino, M. (2021). Computing the original ebwt
faster, simpler, and with less memory. In SPIRE, pages
129–142. Springer International Publishing.
Burrows, M. and Wheeler, D. (1994). A Block Sorting data
Compression Algorithm. Technical report, DIGITAL
System Research Center.
Chandak, S., Tatwawadi, K., Ochoa, I., Hernaez, M.,
and Weissman, T. (2018). SPRING: a next-
generation compressor for FASTQ data. Bioinformat-
ics, 35(15):2674–2676.
Cleary, J. and Witten, I. (1984). Data compression using
adaptive coding and partial string matching. IEEE
Transactions on Communications, 32(4):396–402.
Cox, A., Bauer, M., Jakobi, T., and Rosone, G.
(2012). Large-scale compression of genomic se-
quence databases with the Burrows-Wheeler trans-
form. Bioinformatics, 28(11):1415–1419.
Deorowicz, S. (2020). Fqsqueezer: k-mer-based compres-
sion of sequencing data. Scientific reports, 10(1):1–9.
DePristo, M. A. and et al. (2011). A framework for variation
discovery and genotyping using next-generation DNA
sequencing data. Nature genetics, 43(5):491–498.
Egidi, L., Louza, F. A., Manzini, G., and Telles, G. P.
(2019). External memory BWT and LCP computation
for sequence collections with applications. Algorithms
for Molecular Biology, 14(1):6:1–6:15.
Ferragina, P. and Manzini, G. (2000). Opportunistic data
structures with applications. In FOCS, pages 390–
398. IEEE Computer Society.
Gagie, T., Navarro, G., and Prezza, N. (2020). Fully Func-
tional Suffix Trees and Optimal Text Searching in
BWT-Runs Bounded Space. J. ACM, 67(1):2:1–2:54.
Greenfield, D. L., Stegle, O., and Rrustemi, A. (2016).
GeneCodeq: quality score compression and improved
genotyping using a Bayesian framework. Bioinfor-
matics, 32(20):3124–3132.
Guerrini, V., Louza, F., and Rosone, G. (2020). Metage-
nomic analysis through the extended Burrows-
Wheeler transform. BMC Bioinformatics, 21.
Hach, F., Numanagi
´
c, I., Alkan, C., and Sahinalp, S. C.
(2012). SCALCE: boosting sequence compression al-
gorithms using locally consistent encoding. Bioinfor-
matics, 28(23):3051–3057.
Hernaez, M., Pavlichin, D., Weissman, T., and Ochoa, I.
(2019). Genomic data compression. Annual Review
of Biomedical Data Science, 2(1):19–37.
Lossy Compressor Preserving Variant Calling through Extended BWT
47

Janin, L., Rosone, G., and Cox, A. J. (2014). Adap-
tive reference-free compression of sequence quality
scores. Bioinformatics, 30(1):24–30.
Kimura, K. and Koike, A. (2015). Ultrafast SNP analysis
using the Burrows-Wheeler transform of short-read
data. Bioinformatics, 31(10):1577–1583.
Li, H. (2013). Aligning sequence reads, clone se-
quences and assembly contigs with BWA-MEM.
arXiv:1303.3997.
Li, H. (2014). Toward better understanding of artifacts in
variant calling from high-coverage samples. Bioinfor-
matics, 30(20):2843–2851.
Li, H. and Durbin, R. (2010). Fast and accurate long-read
alignment with Burrows-Wheeler transform. Bioin-
formatics, 26(5):589–595.
Louza, F. A., Telles, G. P., Gog, S., Prezza, N., and Rosone,
G. (2020). gsufsort: constructing suffix arrays, lcp
arrays and bwts for string collections. Algorithms for
Molecular Biology, 15.
Malysa, G., Hernaez, M., Ochoa, I., Rao, M., Ganesan, K.,
and Weissman, T. (2015). QVZ: lossy compression of
quality values. Bioinformatics, 31(19):3122–3129.
Manber, U. and Myers, G. (1990). Suffix arrays: A new
method for on-line string searches. In ACM-SIAM
SODA, pages 319–327.
Mantaci, S., Restivo, A., Rosone, G., and Sciortino, M.
(2007). An extension of the Burrows-Wheeler Trans-
form. Theoret. Comput. Sci., 387(3):298–312.
Mantaci, S., Restivo, A., Rosone, G., and Sciortino, M.
(2008). A new combinatorial approach to sequence
comparison. Theory Comput. Syst., 42(3):411–429.
Moffat, A. (1990). Implementing the ppm data compres-
sion scheme. IEEE Transactions on Communications,
38(11):1917–1921.
Numanagi
´
c, I., Bonfield, J., Hach, F., Voges, J., and Sahi-
nalp, C. (2016). Comparison of high-throughput se-
quencing data compression tools. Nature Methods,
13:1005–1008.
Ochoa, I., Hernaez, M., Goldfeder, R., Weissman, T., and
Ashley, E. (2016). Effect of lossy compression of
quality scores on variant calling. Briefings in Bioin-
formatics, 18(2):183–194.
Prezza, N., Pisanti, N., Sciortino, M., and Rosone, G.
(2019). SNPs detection by eBWT positional cluster-
ing. Algorithms for Molecular Biology, 14(1):3.
Prezza, N., Pisanti, N., Sciortino, M., and Rosone, G.
(2020). Variable-order reference-free variant discov-
ery with the Burrows-Wheeler transform. BMC Bioin-
formatics, 21.
Prezza, N. and Rosone, G. (2021). Space-efficient construc-
tion of compressed suffix trees. Theoretical Computer
Science, 852:138 – 156.
Roguski, L., Ochoa, I., Hernaez, M., and Deorowicz, S.
(2018). FaStore: a space-saving solution for raw se-
quencing data. Bioinformatics, 34(16):2748–2756.
Shibuya, Y. and Comin, M. (2019). Better quality score
compression through sequence-based quality smooth-
ing. BMC Bioinform., 20-S(9):302:1–302:11.
Yu, Y. W., Yorukoglu, D., Peng, J., and Berger, B. (2015).
Quality score compression improves genotyping ac-
curacy. Nature biotechnology, 33(3):240—243.
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
48
