
Aggregating Statistically Correlated Metabolic Pathways Into Groups to
Improve Prediction Performance
Abdur Rahman M. A. Basher
1 a
and Steven J. Hallam
1,2 b
1
Graduate Program in Bioinformatics, University of British Columbia, Vancouver, BC V5Z 4S6, Canada
2
Department of Microbiology & Immunology, University of British Columbia, Vancouver, BC V6T 1Z3, Canada
Keywords:
Pathway Group, Relabeling, Data Augmentation, Correlated Models, Metabolic Pathway Prediction,
MetaCyc.
Abstract:
Metabolic pathway prediction from genomic sequence information is an essential step in determining the ca-
pacity of living things to transform matter and energy at different levels of biological organization. A detailed
and accurate pathway map enables researchers to interpret and engineer the flow of biological information
from genotype to phenotype in both organismal and multi-organismal contexts. In this paper, we propose
two novel hierarchical mixture models, SOAP (sparse correlated pathway group) and SPREAT (distributed
sparse correlated pathway group), to improve pathway prediction outcomes. Both models leverage pathway
abundance to represent an organismal genome as a mixed distribution of groups, and each group, in turn, is
a mixture of pathways. Moreover, both models deal with missing potential pathways in the training set by
provisioning supplementary pathways into the learning framework as part of noise reduction efforts. Because
the introduction of supplementary pathways may lead to overestimation of some pathways, dual sparseness is
applied. The resulting pathway group dataset is then used to train multi-label learning algorithms. Model ef-
fectiveness was evaluated on metabolic pathway prediction where correlated models, in particular, SOAP was
able to equal or exceed the performance of previous pathway prediction algorithms on organismal genomes.
1 INTRODUCTION
Rapid advances in high-throughput sequencing and
mass spectrometry over the past two decades have
produced a veritable tidal wave of multi-omic infor-
mation spanning the central dogma of biology (DNA,
RNA, protein and metabolites) at the individual, pop-
ulation and community levels of organization (Wang
et al., 2015) (Hassa et al., 2018) (Aguiar-Pulido et al.,
2016) (Loh et al., 2012). As the ubiquity and abun-
dance of these datasets increases there is a concomi-
tant need to develop bioinformatics applications that
scale with data volume and complexity. In particu-
lar, methods for predicting metabolic pathways have
become essential to interpret and engineer the flow
of biological information from genotype to phenotype
(Lawson et al., 2019) (Hahn et al., 2016).
A metabolic pathway can be defined as a series
of linked chemical reactions occurring within or be-
tween cells, often catalyzed and coordinated by a
a
https://orcid.org/0000-0002-3407-1187
b
https://orcid.org/0000-0002-4889-6876
group of enzymes, resulting in metabolic flux from
substrate to product and so on to completion. A va-
riety of rule-based and machine learning prediction
methods have been developed to model these path-
ways in both organismal and multi-organismal con-
texts (Mascher et al., 2019) (Baranwal et al., 2020)
(Yamanishi et al., 2015) (Tabei et al., 2016) (Ye
and Doak, 2009) (Dale et al., 2010) (Karp et al.,
2016) (M. A. Basher et al., 2020) (M. A. Basher
et al., 2021b). While these methods rely on reference
metabolic pathway databases (e.g., MetaCyc (Caspi
et al., 2019) and KEGG (Kanehisa et al., 2017)) to re-
construct pathways, other computational methods ig-
nore the use of reference database or follow an agnos-
tic approach pathway boundaries in the reconstruction
process (Zhao et al., 2012) (Qi et al., 2014) (Shafiei
et al., 2014) (Jiao et al., 2013).
Among recently developed pathway prediction
methods is triUMPF (M. A. Basher et al., 2021b)
which uses several layers of interactions among path-
ways and enzymes within a network to improve the
precision of pathway predictions in terms of com-
munities represented by a cluster of nodes (pathways
M. A. Basher, A. and Hallam, S.
Aggregating Statistically Correlated Metabolic Pathways Into Groups to Improve Prediction Performance.
DOI: 10.5220/0010910100003123
In Proceedings of the 15th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2022) - Volume 3: BIOINFORMATICS, pages 49-61
ISBN: 978-989-758-552-4; ISSN: 2184-4305
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
49

Figure 1: Group-based pathway prediction workflow. The
training phase (a) takes pathway abundance data to discover
groups using any correlated models in the CHAP pack-
age. Then, groups are used to map examples in the path-
way abundance data to groups using reMap (Hallam Lab,
2021b). Then, the results of this mapping are used in leADS
(Hallam Lab, 2021a), along with pathway data, to learn
the model. After training, pathways can be predicted for
a newly sequenced genome (b), by first inferring groups us-
ing reMap, and then apply the pretrained leADS to predict
pathways from groups.
and enzymes). Despite triUMPF’s predictive gains,
its performance remained error prone because its pre-
diction process depends on the quality of commu-
nities detected from both pathway and enzyme net-
works that are learned from pathway datasets which
contain missing pathway information (M. A. Basher
et al., 2020).
Previously, Shafiei and colleagues developed
BiomeNet (Shafiei et al., 2014), an extension of
MetaNetSim (Jiao et al., 2013), which is a hierarchi-
cal Bayesian network to reconstruct metabolic net-
works in a purely data-driven manner by leverag-
ing enzyme abundances present in multi-organismal
datasets. Instead of relying on defined pathway
boundaries (Khatri et al., 2012), BiomeNet discovers
functions that are referred to as subnetworks, where
a subnetwork constitutes a group of connected reac-
tions. Applications of BiomeNet to the human gut
microbiome revealed subnetworks that are common
among healthy and inflammatory bowel disease (IBD)
microbiome patients as well as distinct subnetworks
associated with IBD patients.
Inspired by BiomeNet, we developed CHAP
(correlated pathway-group) a software package com-
prised of three correlated mixed-membership hier-
archical Bayesian models, CTM (Blei and Lafferty,
2006), SOAP, and SPREAT, to capture mixed compo-
nents given pathway abundance data. The component
is referred to as a “pathway group”, which is com-
prised of a set of correlated pathways, while path-
ways are permitted to be inter-mixed across groups
with different proportions, resulting in overlapping
pathways on groups. Modeling explicitly correlations
among pathways, using a Gaussian covariance matrix,
is fundamental as functions of similar organisms or
communities are shared. Moreover, due to noise or
missing pathways information, the two novel models:
SOAP and SPREAT provision supplementary path-
ways into the learning framework as part of noise
reduction efforts. Because the introduction of sup-
plementary pathways may lead to overestimation of
some pathways, dual sparseness is applied where each
example in the pathway abundance data is represented
by a few focused mixing groups and each pathway
group consists of a few relevant pathways. These last
two properties were not included in CTM. By model-
ing examples as mixing groups, one may use results
from correlated models for downstream group-based
pathway prediction (Fig. 1).
Using CTM, SOAP, and SPREAT, we evaluated
groups on metabolic pathway prediction. Resulting
pathway group datasets were used to train reMap
(Hallam Lab, 2021b) to map examples to groups.
For pathway prediction using groups, we applied
leADS software (Hallam Lab, 2021a) using the rec-
ommended settings discussed in (M. A. Basher and
Hallam, 2021). The results were then compared to
two heuristic or rule-based pathway prediction algo-
rithms: MinPath (Ye and Doak, 2009) and Patho-
Logic (Karp et al., 2016), and to two machine
learning algorithms: mlLGPR (M. A. Basher et al.,
2020) and triUMPF (M. A. Basher et al., 2021a)
on a set of Tier 1 (T1) pathway genome databases
(PGDBs) and genomes used in the Critical Assess-
ment of Metagenome Interpretation (CAMI) initiative
(Sczyrba et al., 2017) following established bench-
marks (M. A. Basher et al., 2020).
2 CORRELATED MODELS
In this section, we present three correlated path-
way models: i)-CTM (correlated topic model) (Blei
and Lafferty, 2006), ii)- SOAP (sparse correlated
pathway group) and iii)- SPREAT (distributed sparse
correlated pathway group). These models incorporate
pathway abundance information to encode each ex-
ample as a mixture distribution of groups, and each
pathway group, in turn, is a mixture of pathways
with different mixing proportions. The pathway abun-
dance information can be obtained by mapping en-
zyme –with abundances– onto a reference pathway
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
50
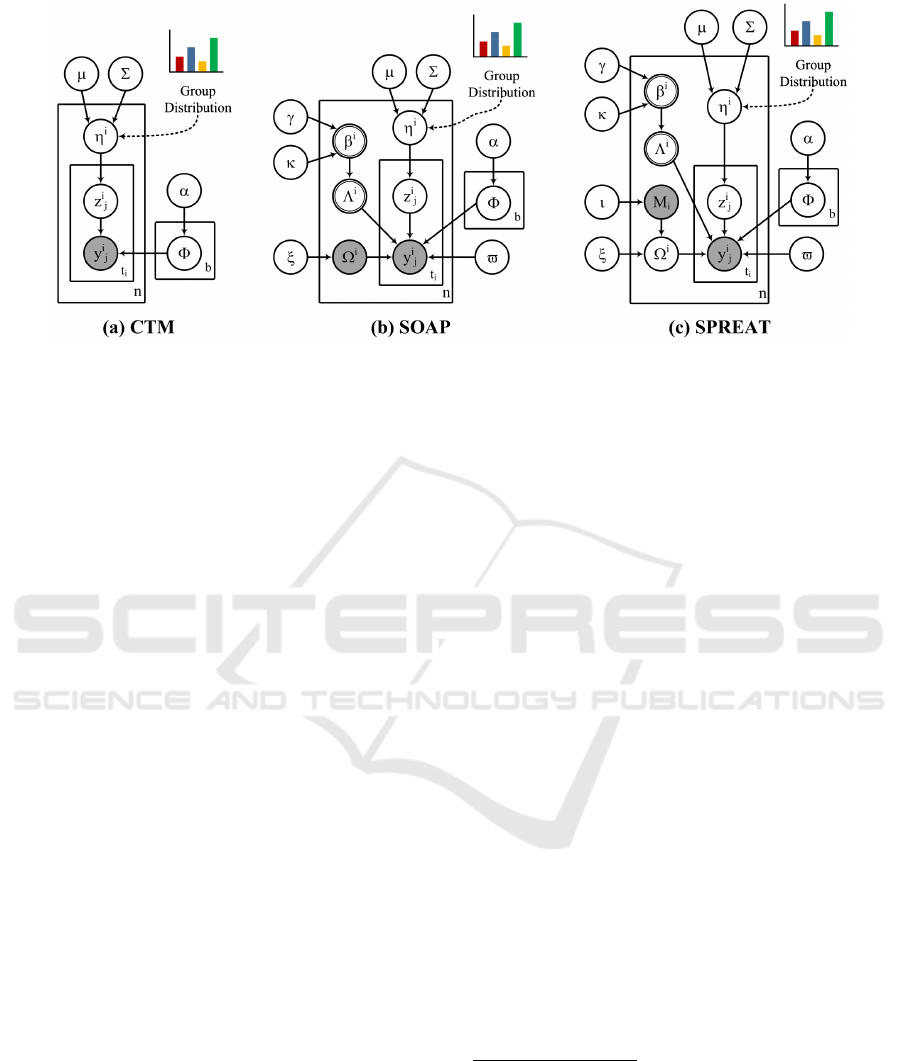
Figure 2: Graphical model representation of the correlated group models. The boxes are “plates” representing replicates. The
outer plate represents examples, while the inner plate represents the repeated choice of pathways for an example. The logistic
normal distribution, used to model the latent group proportions for an example, captures correlations among groups that are
impossible to capture using a single Dirichlet. The observed data for each example i are a set of annotated pathways y
(i)
and a set of hypothetical pathways M
i
. The hidden variables are: per-example group proportions η
(i)
, per-example group
selection parameters Λ
(i)
, per-example hypothetical pathway distributions Ω
(i)
, per-pathway group assignment parameter z
(i)
j
,
and per-group distribution over pathways Φ
a
.
database (e.g. MetaCyc (Caspi et al., 2019)). Be-
fore we discuss these three models, let us first provide
some definitions and notations.
Pathway Abundance Data. Let P = {y
(i)
: 1 <
i 6 n} be a collection of n examples correspond-
ing organismal or multi-organismal genomes (e.g.
Escherichia coli K-12), where each example y
(i)
=
(y
(i)
1
,y
(i)
1
,...,y
(i)
t
) is a vector encoding the unnormal-
ized abundance information of pathways and t is the
pathway size. Let Y = {h
1
,h
2
,...,h
t
} be a set of
all known metabolic pathways obtained from a ref-
erence database (e.g., MetaCyc (Caspi et al., 2019)),
and Y
i
⊆ Y corresponds to a subset of true pathways
associated with the ith example.
Group Modeling. Given P , a pathway group dis-
tribution for the ith example is a multinomial distri-
bution vector, denoted by η
(i)
of size b groups, i.e.,
{p(Φ
a
|η
(i)
)}
a=b
a=1
, where Φ
j
in a multinomial pathway
distribution over the group j, i.e., {p(y
k
|Φ
j
)}
k=t
k=1
. The
overall goal of group modeling is to discover b hidden
groups for each example.
The definition states that a pathway is distributed
over groups, implying group correlation, i.e., if l ∈
Φ
j
≥ 0 and l ∈ Φ
k
> 0 then Σ
j,k
6= 0, where Σ ∈ R
b×b
is a group-correlation matrix.
Group Correlation. Given P , the pairwise group-
correlation is defined by a Gaussian covariance ma-
trix, denoted by Σ ∈ R
b×b
. Each entry s
i, j
in Σ char-
acterizes the magnitude of correlation between i and
j pathway groups, where a larger score indicates both
pathway groups are highly correlated.
Missing pathway information in P is common in
both organismal and multi-organismal contexts due
to errors in open reading frame prediction or anno-
tation as well as unknown protein function. Previ-
ously, Hanson and colleagues (Hanson et al., 2014)
reported missing a set of potential pathways for the
Hawaii Ocean Time-series data (Stewart et al., 2011),
such as tricarboxylic acid cycle (TCA). These missing
pathways have negative implications in group mod-
eling as P , in this case, would be exposed to ex-
treme noise. Although manually incorporating miss-
ing pathways to P may provide a solution to model
groups, this solution has the potential to increase false
discovery pending experimental validation. A good
compromise would be to record missing pathways in a
separate list while keeping the original pathway abun-
dance data intact. Lets us denote M ∈ Z
n×t
≥0
a matrix
holding a set of missing pathways where each entry is
an integer value indicating the abundance of a path-
way for an example. This matrix is called the back-
ground or the supplementary matrix. Now, with these
definitions, let us describe the research problem.
Problem Statement. Given P and M, the objective
is to recover the group distribution η for each exam-
ple such that applying group based metabolic pathway
prediction would recover more accurate pathways for
an organismal or multi-organismal genome.
Aggregating Statistically Correlated Metabolic Pathways Into Groups to Improve Prediction Performance
51

1 for a ∈ {1,.. .,b} do
2 Sample a distribution over pathways
Φ
a
∼ Dir(.|α);
3 for i ∈ {1,.. .,n} do
4 Draw per example group weight
η
(i)
∼ N (.|µ, Σ);
5 Draw group proportions θ
(i)
= softmax(η
(i)
);
6 for j ∈ {1,.. .,t
(i)
} do
7 Sample a group assignment
z
(i)
j
∼ Mult(.|θ
(i)
);
8 Sample a pathway y
(i)
j
∼ Mult(.|Φ
z
(i)
j
);
Algorithm 1: The generative process for CTM given a col-
lection of examples.
2.1 Correlated Topic Model
The correlated topic model (CTM) is a probabilistic
graphical model that extends the generative story of
latent Dirichlet allocation (LDA) (Blei et al., 2003) to
incorporate correlation among groups (or topics in the
original paper). Fig. 2a shows the Bayesian graphi-
cal model for CTM using plate notation. Like LDA,
CTM is composed of a hierarchical Bayesian mixture
model, where features (words in the original paper)
are mixed to constitute groups that are assumed to be
correlated, modeled by a Gaussian covariance matrix.
Note that in this paper, we use the terms feature and
pathway interchangeably.
Formally, let n be the total number of examples in
P , where each example i consists of features, i.e., y
(i)
.
Then, the generative process for CTM is described
as follows. First, we draw a multinomial feature
distribution Φ
a
from a Dirichlet prior α > R
>0
for
each group a ∈ {1, . . . , b}. Then, for each example i,
a Gaussian random variable is drawn η
(i)
∼ N (µ,Σ),
where µ is a b dimensional mean and Σ ∈ R
b×b
is
the covariance matrix. The random variable η
(i)
is
projected onto the probability simplex to obtain the
group distributions θ
(i)
= softmax(η
(i)
), correspond-
ing the logistic-normal distribution, from which a
group indicator z
(i)
j
∈ {1,...,b} is sampled. Finally,
each observed feature j ∈ {1,...,t
i
} is drawn from
the associated feature distribution, indicated by it’s
group assignment z
j
, i.e., y
(i)
j
∼ Φ
z
(i)
j
. This generative
process (Algorithm 1) is identical to LDA except that
the group distributions is sampled from the logistic
normal instead from a Dirichlet prior as in LDA.
2.2 Correlated Pathway-group Model
Correlated pathway group models are extensions to
CTM: i)- SOAP (Fig. 2b) and ii)- SPREAT (Fig. 2c).
Both models incorporate dual sparseness and supple-
mentary pathways in modeling group proportions.
The two properties were not implemented in CTM.
Let us discuss these two models.
Analogous to CTM, given n number of examples
in P and a matrix encoding missing pathways M, the
generative process for SOAP and SPREAT can be
described as follows. First, we draw a multinomial
pathway distribution Φ
a
from asymmetric Dirichlet
prior α ∈ R
>0
for each group a ∈ {1,...,b}, where b
is assumed to be known and fixed in advance. The
symmetric assumption is appropriate, in such a sce-
nario, because our prior knowledge, associated with
these pathways, is inaccessible. For each example
i, a group proportion is drawn θ
(i)
= softmax(η
(i)
),
where η
(i)
is a Gaussian random variable with mean
and covariance are denoted by µ and Σ, respectability.
To sample a group, it is reasonable to expect
that: i)- each example is usually explained with a
handful set of a mixed proportion of groups and ii)- a
group should consist only of a few related pathways.
Therefore, we apply dual sparsity (Lin et al., 2014)
(Airoldi et al., 2008) (Bien and Tibshirani, 2011)
(He et al., 2017) to retain those relevant focused
groups and pathways by: i)- introducing an auxiliary
Bernoulli variable Λ
(i)
of size b to determine whether
a group is selected for the ith example or ignored
and ii)- applying a cutoff threshold to keep top
k t pathways, based on their probabilities, for
each group. Instead of sampling each entry in Λ
(i)
directly from a Bernoulli coin toss, we assume that
each entry is sampled from a Beta distribution β
(i)
,
parameterized by two hyperparameters γ ∈ R
>0
and
κ ∈ R
>0
. Applying this dual sparsity, we aim to
enhance the interpretability of the learned pathway
groups while reducing the negative correlation among
groups on Σ.
Next, a group indicator z
(i)
j
∈ {1,...,b} is drawn
according to the example-specific mixture proportion
Λ
(i)
θ
(i)
, where represents the Hadamard prod-
uct. Now each pathway y
(i)
j
for the ith example is
generated from a weighted distribution Ω
(i)
z
(i)
j
Φ
z
(i)
j
using a smoothing prior ϖ ∈ R
>0
. The parameter
Ω
(i)
∈ R
t
, derived from M
i
, represents a normal-
ized supplementary pathway of size t, which is as-
sumed to be drawn from a symmetric Dirichlet prior
ξ ∈ R
>0
. For SPREAT, this parameter encodes distri-
bution, where each element of Ω
(i)
j
corresponds to the
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
52
pathway probability y
(i)
j
∈ M
i
for ith example. Here,
the background pathway is assumed to be drawn from
a sparse binary vector prior ι ∈ R
>0
that is included
for completeness because pathways in M for each ex-
ample are known.
1 for a ∈ {1,.. .,b} do
2 Sample a distribution over pathways
Φ
a
∼ Dir(.|α);
3 for i ∈ {1,.. .,n} do
4 Draw per example group weight
η
(i)
∼ N (.|µ, Σ);
5 Draw group proportions θ
(i)
= softmax(η
(i)
);
6 Draw beta distribution β
(i)
∼ Beta(.|γ,κ);
7 Draw a sparsity indicator vector
Λ
(i)
∼ Bernoulli(.|β
(i)
);
8 if SPREAT then
9 Sample a vector M
i
∼ Prior(.|ι);
10 Sample background distribution
Ω
(i)
|M
i
∼ Dir(.|ξ);
11 else
12 Draw background feature proportions
Ω
(i)
∼ Dir(.|ξ);
13 for j ∈ {1,.. .,t
(i)
} do
14 Sample a group assignment
z
(i)
j
∼ Mult(.|Λ
(i)
θ
(i)
);
15 Sample a pathway
y
(i)
j
∼ Mult(.|(1 − Ω
(i)
z
(i)
j
) Φ
z
(i)
j
);
Algorithm 2: The generative process for SOAP and
SPREAT.
Representing SOAP and SPREAT as layer-wise
mixing components supports the hierarchical mod-
ularity of metabolic pathway generation, where the
components of one level (e.g., pathways) permit to
contribute to groups with different degrees of granu-
larity. The generative process of SOAP and SPREAT
models is summarized in Algorithm 2. Notice that
by setting all entries in Ω, Λ, and ϖ to 1, SOAP
and SPREAT are reduced to CTM (“collapse2ctm” or
c2m), reflecting the flexibility of these models.
3 INFERENCE AND PARAMETER
ESTIMATION FOR SPREAT
Here, we discuss the inference for the SPREAT
model. A similar expression is derived for SOAP.
Given P , the goal of inference is to compute the
posterior distribution of per-example group propor-
tions (η), per-example group selection parameters (Λ)
Table 1: Correspondence between variational and original
parameters.
Original parameter Φ µ Σ Λ Ω z
Variational parameter φ ν ζ
2
λ ω ς
and the associated beta distributions (β), per-example
background pathway distributions (Ω), per-pathway
group assignment (z), and per-group distribution over
pathways (Φ). By denoting all parameters as Θ and
variables as V while omitting hyperparameters, we
apply the Jensen’s inequality on a variational distri-
bution over hidden variables q(Θ,V) to obtain the ev-
idence lower bound (ELBO) as:
L(q) = E
q
[log p(Y,M, Θ, V)] + H(q)
(3.1)
where p(Y, M, Θ, V) represents the joint distri-
bution of all observed and latent variables of the
model. The ELBO contains two terms. The first term,
E
q
[log p(Y,M, Θ, V)], captures how well q(Θ,V) de-
scribes a distribution of the model. The second
term is the entropy of the variational distribution,
E
q
[−logq(Θ,V)], which protects the variational dis-
tribution from “overfitting”. The two terms depends
on q(Θ,V) which is defined as:
q(Θ,V) =
b
∏
a=1
q(Φ
a
|φ
a
)
"
n
∏
i=1
q(η
(i)
|ν,ζ
2
)
× q(Λ
(i)
|λ
(i)
)q(Ω
(i)
|ω
(i)
)
j=t
i
∏
j=1
q(z
(i)
j
|ς
(i)
j
)
#
(3.2)
where φ,ν,ζ
2
,λ,ω and ς are variational free pa-
rameters. Table 1 shows the correspondence between
variational and the original parameters. Now, the first
term in Eq. 3.1 is decomposed into:
E
q
[log p(Y, M,Θ,V )] =
a=b
∑
a=1
E
q
[log p(Φ
a
|α)]
+
i=n
∑
i=1
E
q
[log p(η|µ, Σ)] + E
q
[log p(Λ
(i)
|β
i
)]
+ E
q
[log p(β
i
|γ,κ)] + E
q
[log p(Ω
(i)
|M
(i)
,ξ)]
+
j=t
i
∑
j=1
E
q
[log p(y
(i)
j
|z
(i)
j
,Ω
(i)
j
,Λ
(i)
,Φ, ϖ)]
+ E
q
[p(z
(i)
j
|η)]
!
(3.3)
where,
E
q
[log p(Φ
a
|α)] = log Γ
j=t
∑
j=1
α
j
−
j=t
∑
j=1
logΓ(α
j
)
Aggregating Statistically Correlated Metabolic Pathways Into Groups to Improve Prediction Performance
53

+
j=t
∑
j=1
(α
j
− 1)E
q
[logΦ
a, j
]
E
q
[log p(η|µ, Σ)] =
1
2
log|Σ
−1
| −
b
2
log2π
−
1
2
tr(diag(ζ
2
)Σ
−1
)
+ (ν −µ)
>
Σ
−1
(ν − µ)
E
q
[log p(Λ
(i)
|β
(i)
)] =
a=b
∑
a=1
λ
(i)
a
logβ
(i)
a
+ (1 −λ
(i)
a
)
× log(1 −β
(i)
a
)
E
q
[log p(β
(i)
|γ,κ))] =
a=b
∑
a=1
(γ − 1)log(β
(i)
a
)
+ (κ −1)log(1 − β
(i)
a
) − log(B(γ,κ)
E
q
[log p(Ω
i
|M
(i)
,ξ)] = log Γ
j=t
∑
j=1
ξ
j
+ M
(i)
j
−
j=t
∑
j=1
logΓ(ξ
j
+ M
(i)
j
)
+
j=t
∑
j=1
(ξ
j
+ M
(i)
j
− 1)E
q
[logΩ
(i)
j
]
E
q
[log p(y
(i)
j
|z
(i)
j
,Ω
(i)
j
,Λ
(i)
,Φ, ϖ)] = log ϖ
+
c=t
∑
c=1
a=b
∑
a=1
y
(i)
j,c
ς
(i)
a, j
λ
(i)
a
E
q
[(1 − Ω
(i)
c
)]E
q
[logΦ
a, j
]
E
q
[log p(z
(i)
j
|η)] ≈ 1 − logρ +
a=b
∑
a=1
ν
a
ς
(i)
a, j
−
k=b
∑
k=1
E
q
[exp(η
k
)]
ρ
−1
The second term H(q) in Eq. 3.1 has the following
parametric forms (see Eq. 3.2):
H(q) = −
a=b
∑
a=1
E
q
[logq(Φ
a
|φ
a
)] −
i=n
∑
i=1
E
q
[logq(η
(i)
|ν,ζ
2
)]
+ E
q
[logq(Λ
(i)
|λ
(i)
)] + E
q
[logq(Ω
(i)
|ω
(i)
)]
+
j=t
i
∑
j=1
E
q
[logq(z
(i)
j
|ς
(i)
j
)]
!
(3.4)
where,
E
q
[logq(Φ
a
|φ
a
)] = log Γ
j=t
∑
j=1
φ
a, j
−
j=t
∑
j=1
logΓ(φ
a, j
)
+
j=t
∑
j=1
(φ
a, j
− 1)E
q
[logΦ
a, j
]
E
q
[logq(η
(i)
|ν,ζ
2
)] = −
a=b
∑
a=1
1
2
logζ
2
a
+ log(2π) +1
E
q
[logq(Λ
(i)
|λ
(i)
)] =
a=b
∑
a=1
λ
(i)
a
logλ
(i)
a
+ (1 −λ
(i)
a
)log(1 − λ
(i)
a
)
E
q
[logq(Ω
(i)
|ω
(i)
)] = log Γ
j=t
∑
j=1
ω
(i)
j
−
j=t
∑
j=1
logΓ(ω
(i)
j
)
+
j=t
∑
j=1
(ω
(i)
j
− 1)E
q
[logΩ
(i)
j
]
E
q
[logq(z
(i)
j
|ς
(i)
j
)] = E
q
h
log
a=b
∏
a=1
(ς
(i)
a, j
)
z
(i)
a, j
i
=
a=b
∑
a=1
ς
(i)
a, j
logς
(i)
a, j
The exceptions that correspond to the above equations
are:
E
q
[logΦ
a, j
] =
Ψ(φ
a, j
) − Ψ(
k=t
∑
k=1
φ
a,k
)
E
q
[logΩ
(i)
j
] =
Ψ(ω
(i)
j
) − Ψ(
k=t
∑
k=1
ω
(i)
k
)
E
q
[(1 − Ω
(i)
c
)] =
1 − ω
(i)
c
∑
k=t
k=1
(1 − ω
(i)
k
)
E
q
[exp(η
k
)] = exp(ν
a
+
1
2
ζ
2
a
)
B(γ,κ) =
Γ(γ)Γ(κ)
Γ(γ + κ)
where Γ denotes the Gamma function while Ψ is the
logarithmic derivative of the Gamma function.
After expanding both terms in Eq. 3.1, we can
now maximize the bound in Eq. 3.1 with respect to
each variational parameters using mini-batch coordi-
nate ascent updates (Hoffman et al., 2013) as:
Optimize ς. The analytical expression of the varia-
tional group assignment q(ς) for each pathway j and
group a for the ith example is not amenable due to
the non-conjugacy of logistic-normal with latent vari-
ables. Instead, we approximate the solution as:
ς
(i)
a, j
∝ exp
c=t
∑
c=1
y
(i)
j,c
λ
(i)
a
1 − ω
(i)
c
∑
k=t
k=1
(1 − ω
(i)
k
)
Ψ(φ
a, j
)
− Ψ(
k=t
∑
k=1
φ
a,k
)
+ ν
a
− 1
!
(3.5)
where Ψ(.) is the digamma function. Notice that
the variational parameter ω
(i)
∗
acts as an smoothing pa-
rameter to selecting groups for each pathways, either
from M
i
or from P .
Optimize ν. Collecting terms in the ELBO bound
that contain only ν and taking derivatives w.r.t. ν
a
for
each group a, we obtain:
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
54

∂L(q)
[ν]
∂ν
a
= − Σ
−1
(ν − µ)+
j=t
i
∑
j=1
ς
(i)
a, j
−
exp(ν
a
+
1
2
ζ
2
a
)
t
i
ρ
−1
(3.6)
where ρ is another variational parameter, as in
CTM (Blei and Lafferty, 2006). The above equation
in hard to optimize, instead, we use a conjugate gra-
dient algorithm.
Optimize ζ
2
. By symmetry, we gather all the terms
that has ζ
2
from Eq. 3.1, and take derivatives w.r.t. ζ
2
a
for each group a to obtain:
∂L(q)
[ζ
2
]
∂ζ
2
a
= −
1
2
Σ
−1
a,a
+t
i
ρ
−1
exp
ν
a
+
1
2
ζ
2
a
−
1
ζ
2
a
(3.7)
Again, there is no analytical solution to the above
formula. Instead, we use Newton’s method for each
coordinate such that ζ
a
∈ R
>0
.
Optimize ρ. We extract terms involved with the vari-
ational parameter ρ, and equating it’s derivative to
zero, we get:
ρ =
k=b
∑
k=1
exp(ν
k
+
1
2
ζ
2
k
)
(3.8)
1 Initialize φ, ν, ζ
2
, λ, ω, ς, γ, κ, ξ, α, ϖ, ι, s = 0,
l ≥ 0, g ∈ (0.5,1]
2 repeat
3 s = s + 1;
4 example a minibatch randomly B ⊂ P ;
5 for i ∈ B do
6 repeat
7 Update ς
(i)
with Eq. 3.5;
8 Update ν
(i)
with Eq. 3.6 using
conjugate gradient algorithm;
9 Update ζ
2,(i)
with Eq. 3.7 using
Newton’s method;
10 Update ρ
(i)
with Eq. 3.8;
11 Update ω
(i)
with Eq. 3.9;
12 Update λ
(i)
with Eq. 3.10;
13 until local variational parameters
converge;
14 Compute optimal values µ =
ν
|B|
,
Σ = diag(
ζ
2
|B|
) + µµ
>
;
15 Compute global optimal values φ with Eq.
3.11;
16 Update the current estimate of the global
variational paramters,
x = (1 − τ)x +τx, where x ∈ {φ,µ,Σ};
17 Update the learning rate τ = (s + l)
−g
;
18 until global convergence criterion is satisfied;
Algorithm 3: Minibatch variational inference for SPREAT.
Optimize ω. We next isolate only the terms in the
bound that contain variational background pathway
distributions q(ω). However, setting it’s derivatives to
zero does not lead to a closed-form solution, instead,
we approximate ω
(i)
c
for each example i according to:
ω
(i)
c
∝ξ
c
+ M
(i)
c
−
1 − ω
(i)
c
−
∑
k=t
k=1
(1 − ω
(i)
k
)
(
∑
k=t
k=1
(1 − ω
(i)
k
))
2
×
j=t
i
∑
j=1
a=b
∑
a=1
y
(i)
j,c
ς
(i)
a, j
λ
(i)
a
Ψ(φ
a, j
) − Ψ(
k=t
∑
k=1
φ
a,k
)
(3.9)
Optimize λ. To optimize λ, we use the canonical pa-
rameterisation of the Bernoulli distribution to get the
following updates for each group a for each example:
λ
(i)
a
=
1
1 + exp
−(log(β
(i)
a
)−log(1−β
(i)
a
))
(3.10)
Optimize φ. Finally, the optimal solution of the vari-
ational pathway distribution q(Φ
a
|φ
a
) for each group
a is obtained by isolating terms involved in the ELBO
bound in Eq. 3.1 and setting it’s gradient to zero:
φ
a,c
=α
c
+
i=n
∑
i=1
j=t
i
∑
j=1
y
(i)
j,c
ς
(i)
a, j
λ
(i)
a
1 − ω
(i)
c
∑
k=t
k=1
(1 − ω
(i)
k
)
(3.11)
The variational inference algorithm samples a
mini-batch from a collection, and uses it to compute
the local latent parameters in Eqs 3.5, 3.6, 3.7, 3.8,
3.9, and 3.10 until the evidence lower bound in Eq.
3.1 converges. Then, the global variational parame-
ter φ is updated in Eq. 3.11 using the posteriors (β,
Λ, η, z, Ω) collected from the previous step after be-
ing scaled according to the learning rate τ = (s+l)
−g
,
where s is the current step, l ≥ 0 is the delay factor,
and g ∈ (0.5, 1] is the forgetting rate. This process for
SPREAT is summarized in Algorithm 3.
3.1 Posterior Predictive Distribution
The posterior predictive distribution estimates the dis-
tribution of an unobserved value (
˜
y) given the ob-
served values (Y
obs
) and parameters (Θ and V) that
are trained on a held-out training set (Hoffman et al.,
2013). The predictive distribution for SPREAT is:
p(
˜
y|Y
obs
,
˜
M,M
obs
) =
Z
p(
˜
y|Θ,
˜
M)p(Θ|Y
obs
,M
obs
)dΘ
≈
a=b
∑
a=1
η
(i)
a
×
j=t
∑
j=1
Φ
a, j
×
˜
y
(i)
j
× q(Θ,V)
(3.12)
where
˜
M is
˜
y’s background pathways and q(Θ,V)
corresponds to Eq. 3.2, trained on Y
obs
and M
obs
.
Aggregating Statistically Correlated Metabolic Pathways Into Groups to Improve Prediction Performance
55
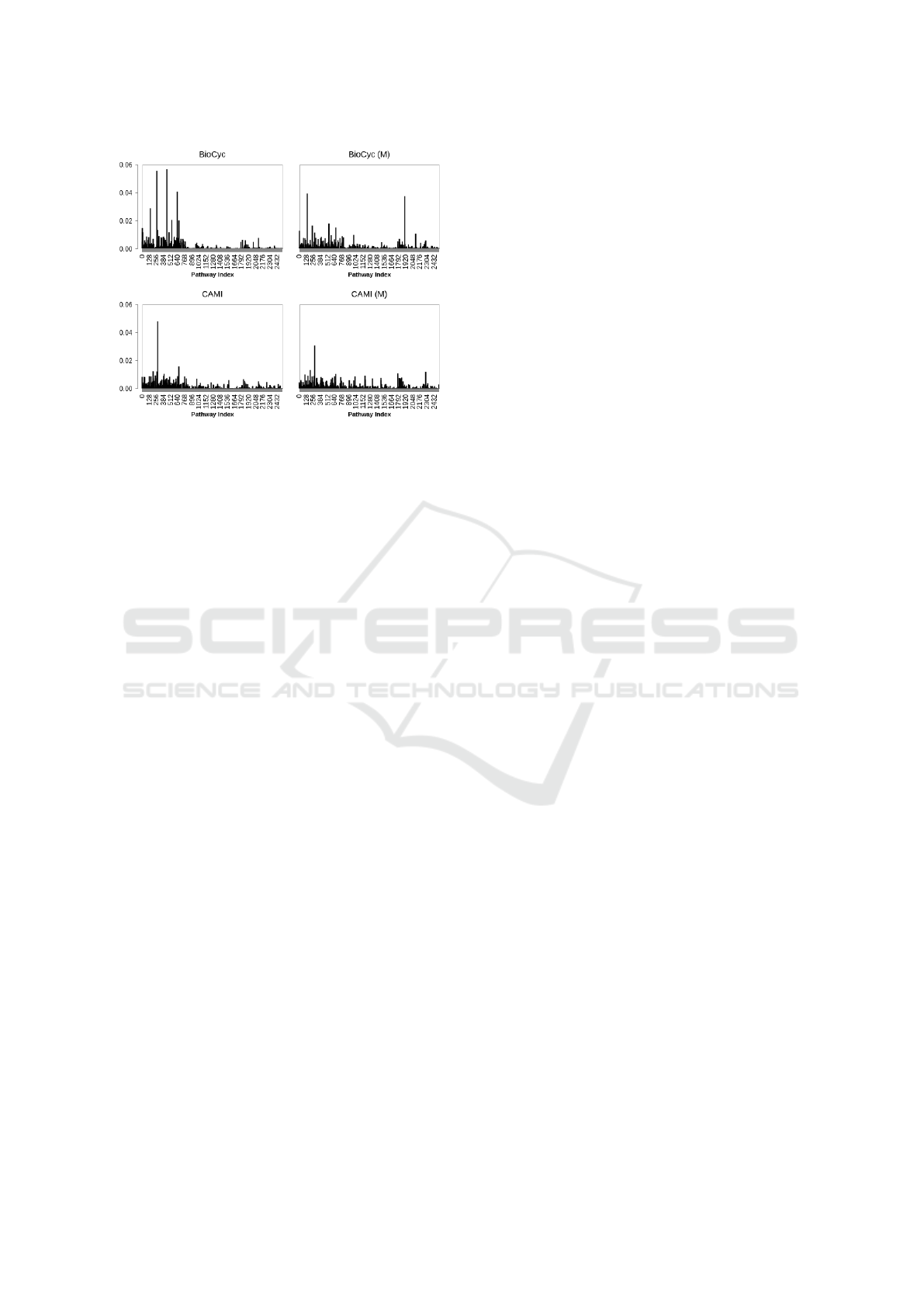
Figure 3: Pathway frequency (averaged on all examples)
in BioCyc (v20.5 T2 &3) and CAMI data, and their back-
ground pathways, indicated by M.
4 EXPERIMENTAL SETTINGS
In this section, we describe the experimental datasets
and settings used to validate the performance of the
three correlated models. The CHAP package was
written in Python v3 and is available under the GNU
license at github.com/hallamlab/chap. All tests were
conducted on a Linux server using 10 cores of Intel
Xeon CPU E5-2650.
4.1 Description of Datasets
The three models were evaluated on diverse path-
way datasets traversing the genomic information hi-
erarchy (M. A. Basher et al., 2020): i)- T1 golden
consisting of EcoCyc, HumanCyc, AraCyc, Yeast-
Cyc, LeishCyc, and TrypanoCyc; ii)- BioCyc (v20.5
T2 & 3) (Caspi et al., 2016); iii)- Critical Assess-
ment of Metagenome Interpretation (CAMI) dataset
composed of 40 genomes (Sczyrba et al., 2017);
and iv)- Synset-2, a noisy training dataset, intro-
duced in (M. A. Basher et al., 2020). For train-
ing, we applied BioCyc (v20.5 T2 & 3) data, while
for evaluating and testing we used T1 golden and
CAMI data. The Synset-2 data was used to ob-
tain supplementary pathways (see Section 4.2). The
preprocessed experimental datasets can be obtained
from zenodo.org/record/5630322#.YYXur2DMK3B
while information about these data is provided in
(M. A. Basher et al., 2020).
4.2 Parameter Settings
Three experiments were conducted: i)- parameter
sensitivity analysis, ii)- groups visualization, and
iii)- metabolic pathway prediction. Unless otherwise
mentioned, we applied the following default configu-
rations: the pathway distribution over groups Φ were
initialized using gamma distribution (with shape and
scale parameters were fixed to 100 and 1/100, re-
spectively), the forgetting rate was g = 0.9, the delay
rate was l = 1, the batch size was 100, the number of
epochs was 3, the number of groups was b = 200, top
k pathways was 100 (only for SOAP and SPREAT),
the Dirichlet hyperparameters α and ξ were 0.0001,
and the beta hyperparameters γ and κ were 2 and 3,
respectively. The supplementary pathways M for Bio-
Cyc, CAMI, and golden T1 datasets were obtained us-
ing mlLGPR (elastic-net with enzymatic reaction and
pathway evidence features)(M. A. Basher et al., 2020)
trained on Synset-2. A schematic view of pathway
frequency for BioCyc T2 &3 and CAMI data with
their background pathways is depicted in Fig. 3.
After obtaining groups, we followed the pathway
prediction pipeline in Fig. 1 by first mapping ex-
amples to groups using reMap software (Hallam Lab,
2021b) and, then, the pathway prediction is achieved
using leADS software (Hallam Lab, 2021a). All hy-
perparameters in reMap, leADS, and mlLGPR, were
fixed to their default values.
5 EXPERIMENTAL RESULTS
This section analyzes the three models using the set-
tings explained in the previous section.
5.1 Sensitivity Analysis
Experimental Setup. Following the common prac-
tice, here we study the effect of hyperparameters on
the performance of correlated models. First, we com-
pare the sensitivity of SOAP and SPREAT against
CTM by incorporating the background pathways M
while varying the number of groups according to
b ∈ {50, 100, 150, 200, 300}. Next, we examine the
SOAP and SPREAT with collapsed option (or c2m)
to compare their performances to CTM, where the
former models should exhibit similar performances
as CTM. Finally, we conduct sparsity analysis of
group distribution by varying the cutoff threshold
value according to k ∈ {50,100,150,200,300,500}
(Section 2.2). For comparative analysis, we apply
CAMI as test data to report the log predictive dis-
tribution (Section 3.1), where a lower score entails
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
56
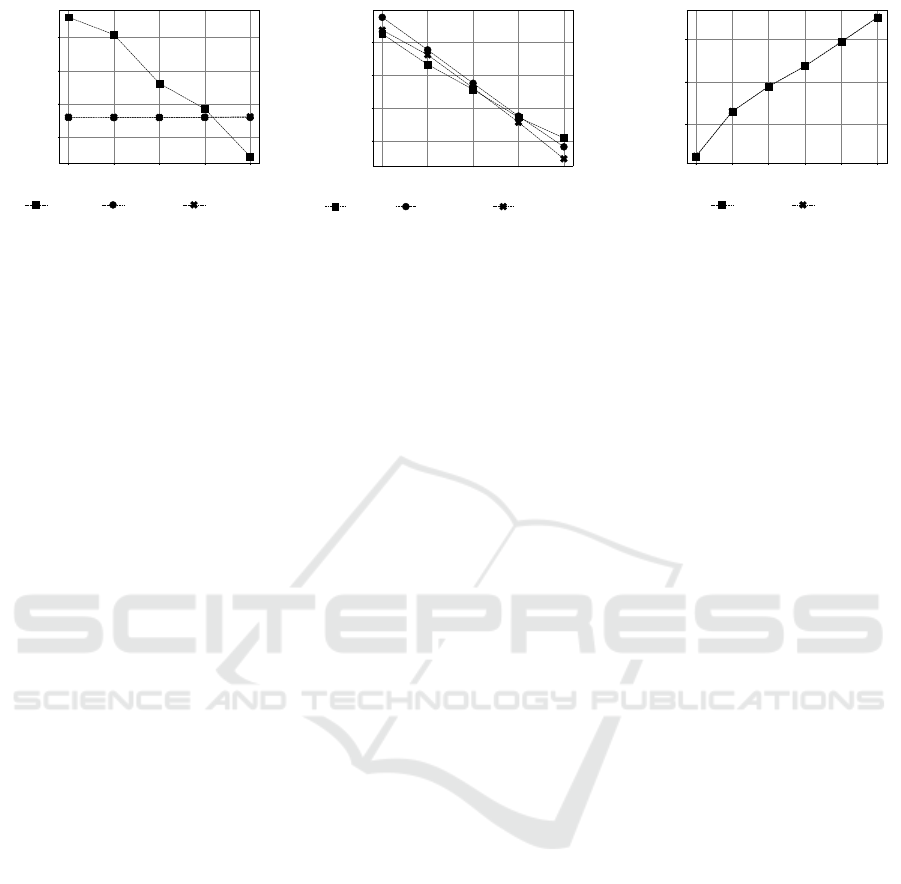
50 100 150 200 300
b
−0.9
−0.8
−0.7
−0.6
Log Pred ic tive Probability
CTM SOAP SPREAT
(a) Effect of b
50 100 150 200 300
b
−0.9
−0.8
−0.7
−0.6
Log Pred ic tive Probability
CTM SOAP+c2m SPREAT+c2m
(b) Effect of b by collapsing
50 100 150 200 300 500
k
−1.0
−0.8
−0.6
Log Pred ic tive Probability
SOAP SPREAT
(c) Effect of k with b = 200
Figure 4: Log predictive distribution on CAMI data. Figs 4a and 4b show the effect of group size b to the performance of
CTM, SOAP, and SPREAT and the collapsed models, respectively. Fig. 4c demonstrates the effect of retaining top k pathways
to the performance SOAP and SPREAT. The performance is measures according to the log predictive probability where higher
values indicate better performances.
higher generalization capability for the corresponding
model.
Experimental Results. While the log predictive
scores for SOAP and SPREAT in Fig. 4a appear to
be flat across various group sizes, the CTM model
projects a more realistic view where its performances
are seen to be gaining by including more groups. Both
SOAP and SPREAT incorporate supplementary path-
ways in modeling the pathway distribution. There-
fore, it is expected to learn additional pathways from
M that has an average of ∼ 500 pathways in relation
to BioCyc v20.5 T2 & 3 which has ∼ 195 pathways
on average. By excluding M (“c2m” in Section 2.2)
in the SOAP and SPREAT training, the log predic-
tive distribution of these models exhibit similar per-
formance as CTM (Fig. 4b). From Figs 4a and 4b, it
is evident that b = 200 represents an optimum group
size. To find an optimum k value, we fixed b = 200
and retrained all models. From Fig. 4c, the perfor-
mances for SOAP and SPREAT are seen to decline
(< −0.6) when k > 100.
Results from this experiment suggest that the set-
tings b ∈ Z
[150,300]
and k ∈ Z
[50,100]
are optimum for
discovering pathway groups in P .
5.2 Groups Visualization
Experimental Setup. Recall that groups constitute
overlapping pathways. In this experiment, we visu-
ally explore the recovered groups from the three cor-
related models trained on BioCyc (v20.5 T2 &3) data
using configurations discussed in Section 4.2. We in-
vestigate group correlations, reflected in Σ, for SOAP,
SPREAT, CTM, SOAP+c2m, and SPREAT+c2m
models, to analyze the influence of dual sparseness
(Section 2.2) and background pathways on Σ.
Experimental Results. Fig. 5 demonstrates 50 ran-
domly picked groups and their correlations as rep-
resented by Σ for all models. The width of edges
indicates the strength of correlations. Essentially
for every group in these models, there are approxi-
mately 12 to 19 closely related groups. This indicates
that metabolic pathways are distributed over multi-
ple groups, therefore, forming overlapping pathways.
With regard to M, as explained in Section 5.1, back-
ground pathways in M consist of ∼ 500 pathways for
organismal or multi-organismal genomes in compari-
son to BioCyc (v20.5 T2 &3) data that has an average
of ∼ 195 pathways. These additive pathways influ-
ence the construction of group correlation for both
SOAP and SPREAT. Pathway groups in SOAP con-
sist of more associated groups (∼ 19 groups) than the
remaining models. This has an important implication
for pathway prediction outcomes, discussed in Sec-
tion 5.3. Sparse models share a similar group struc-
ture as CTM (also they have similar log predictive
scores in Section 5.1), therefore, they may exhibit
similar effects on pathway prediction performance.
Results from this experiment show that SOAP and
SPREAT are better contenders than CTM. Specifi-
cally, both models incorporate supplementary path-
ways and apply dual sparseness to reduce both the
group size and the statistically irrelevant pathways.
5.3 Metabolic Pathway Prediction
Experimental Setup. Pathway groups obtained from
correlated models are used for pathway prediction.
We consider five models: CTM, two models with
background pathways (SOAP and SPREAT), and two
collapsed models (SOAP+c2m and SPREAT+c2m).
After obtaining groups, we trained reMap using the
configuration discussed in Section 4.2. The results
are reported on golden T1 data using four evaluation
metrics: Hamming loss, average precision, average
Aggregating Statistically Correlated Metabolic Pathways Into Groups to Improve Prediction Performance
57

(a) SOAP (#groups:
∼ 19)
(b) SPREAT
(#groups: ∼ 13)
(c) CTM (#groups:
∼ 12)
(d) SOAP+c2m
(#groups: ∼ 12)
(e) SPREAT+c2m
(#groups: ∼ 12)
Figure 5: 50 randomly picked pathway groups, represented by blue circles, and their correlations, indicated by black links,
for each model. The average number of related groups to each pathway group is indicated by #groups. CTM, SOAP+c2m,
and SPREAT+c2m form two distinct clusters of groups, indicating pathways are less shared among groups while SOAP and
SPREAT have more shared pathways in their groups.
recall, and average F1 score. For comparative
analysis, four pathway prediction algorithms are
used: i)- MinPath v1.2 (Ye and Doak, 2009), ii)-
PathoLogic v21 (Karp et al., 2016), iii)- mlLGPR
(elastic net with enzymatic reaction and pathway
evidence features) (M. A. Basher et al., 2020), and
iv)- triUMPF (M. A. Basher et al., 2021a).
Experimental Results. Table 2 shows that groups
from SOAP result in competitive performance against
the other methods in terms of average F1 score with
optimal performance on EcoCyc (0.8336). However,
it seems to be underperforming on AraCyc, YeastCyc,
and LeishCyc, yielding average F1 scores of 0.4764,
0.4914, and 0.4144, respectively. This is attributed
to incorrect background pathways in M (see Section
5.1), hence, impacting the training process. Inter-
estingly, SPREAT’s performance appears to be infe-
rior to SOAP. As alluded in Section 5.2, the average
number of correlated groups for SOAP is significantly
larger than SPREAT (Section 5.2), enforcing to revisit
a true positive pathway for an organism multiple times
across groups in SOAP to signal its presence in con-
trast to groups from SPREAT. With respect to the sen-
sitivity score, correlated models, in general, resulted
in higher scores than triUMPF reinforcing the benefit
of modeling groups to improve predictions.
Results from this experiment demonstrate that the
group-based approach, in particular SOAP, improves
metabolic pathway prediction outcomes. We suggest
applying SOAP for pathway predictions using the de-
fault configurations discussed in Section 4.2.
6 CONCLUSIONS
In this paper, we presented two novel statistical hier-
archical mixture models, SOAP and SPREAT, to un-
cover correlated pathway groups given pathway abun-
dance data. The work is motivated by the prob-
lem of missing pathways, which is very common
in pathway prediction from organismal and multi-
organismal datasets. We empirically evaluated corre-
lated models for pathway prediction using golden T1
data and compared results to other prediction meth-
ods including PathoLogic, MinPath, mlLGPR, and
triUMPF. Overall, correlated models showed promis-
ing results in boosting prediction performance over
ML-based algorithms, such as triUMPF. There are
several directions for future study. Foremost, we in-
tend to build a model that combines both graph-based
(M. A. Basher et al., 2021a) and group-based meth-
ods to improve metabolic pathway prediction with
emphasis on multi-organismal genomes. Additional
attention should be paid to sparseness induction in
the covariance matrix for better interpretability (Fan
et al., 2016).
ACKNOWLEDGEMENTS
This work was performed under the auspices of
Genome Canada, Genome British Columbia, the
Natural Sciences and Engineering Research Council
(NSERC) of Canada, and Compute/Calcul Canada).
ARMAB was supported by a UBC four-year doc-
toral fellowship (4YF) administered through the UBC
Graduate Program in Bioinformatics.
REFERENCES
Aguiar-Pulido, V., Huang, W., Suarez-Ulloa, V., Cickovski,
T., Mathee, K., and Narasimhan, G. (2016). Metage-
nomics, metatranscriptomics, and metabolomics ap-
proaches for microbiome analysis: supplementary is-
sue: bioinformatics methods and applications for big
metagenomics data. Evolutionary Bioinformatics,
12:EBO–S36436.
Airoldi, E. M., Blei, D. M., Fienberg, S. E., and Xing, E. P.
(2008). Mixed membership stochastic blockmodels.
Journal of machine learning research, 9(Sep):1981–
2014.
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
58

Table 2: Predictive performance of each comparing algorithm on 6 benchmark datasets. For each performance metric, ‘↓’
indicates the smaller score is better while ‘↑’ indicates the higher score is better. Bold text suggests the best performance in
each column.
Methods
Hamming Loss ↓
EcoCyc HumanCyc AraCyc YeastCyc LeishCyc TrypanoCyc
PathoLogic 0.0610 0.0633 0.1188 0.0424 0.0368 0.0424
MinPath 0.2257 0.2530 0.3266 0.2482 0.1615 0.2561
mlLGPR 0.0804 0.0633 0.1069 0.0550 0.0380 0.0590
triUMPF 0.0435 0.0954 0.1560 0.0649 0.0443 0.0776
SOAP 0.0392 0.0400 0.1714 0.0934 0.0772 0.0479
SPREAT 0.0519 0.0827 0.1489 0.0748 0.0629 0.0503
CTM 0.0558 0.0835 0.1425 0.0804 0.0622 0.0503
SOAP+c2m 0.0590 0.0780 0.1457 0.0772 0.0614 0.0534
SPREAT+c2m 0.0542 0.0796 0.1520 0.0772 0.0598 0.0558
Methods
Average Precision Score ↑
EcoCyc HumanCyc AraCyc YeastCyc LeishCyc TrypanoCyc
PathoLogic 0.7230 0.6695 0.7011 0.7194 0.4803 0.5480
MinPath 0.3490 0.3004 0.3806 0.2675 0.1758 0.2129
mlLGPR 0.6187 0.6686 0.7372 0.6480 0.4731 0.5455
triUMPF 0.8662 0.6080 0.7377 0.7273 0.4161 0.4561
SOAP 0.8611 0.7871 0.6215 0.4851 0.2805 0.5985
SPREAT 0.9400 0.6750 0.8350 0.6000 0.3200 0.6200
CTM 0.9150 0.6700 0.8750 0.5650 0.3250 0.6200
SOAP+c2m 0.8950 0.7050 0.8550 0.5850 0.3300 0.6000
SPREAT+c2m 0.9250 0.6950 0.8150 0.5850 0.3400 0.5850
Methods
Average Recall Score ↑
EcoCyc HumanCyc AraCyc YeastCyc LeishCyc TrypanoCyc
PathoLogic 0.8078 0.8423 0.7176 0.8734 0.8391 0.7829
MinPath 0.9902 0.9713 0.9843 1.0000 1.0000 1.0000
mlLGPR 0.8827 0.8459 0.7314 0.8603 0.9080 0.8914
triUMPF 0.7590 0.3835 0.3529 0.3319 0.7126 0.6229
SOAP 0.8078 0.8746 0.3863 0.4978 0.7931 0.9371
SPREAT 0.6124 0.4839 0.3275 0.5240 0.7356 0.7086
CTM 0.5961 0.4803 0.3431 0.4934 0.7471 0.7086
SOAP+c2m 0.5831 0.5054 0.3353 0.5109 0.7586 0.6857
SPREAT+c2m 0.6026 0.4982 0.3196 0.5109 0.7816 0.6686
Methods
Average F1 Score ↑
EcoCyc HumanCyc AraCyc YeastCyc LeishCyc TrypanoCyc
PathoLogic 0.7631 0.7460 0.7093 0.7890 0.6109 0.6447
MinPath 0.5161 0.4589 0.5489 0.4221 0.2990 0.3511
mlLGPR 0.7275 0.7468 0.7343 0.7392 0.6220 0.6768
triUMPF 0.8090 0.4703 0.4775 0.4735 0.5254 0.5266
SOAP 0.8336 0.8285 0.4764 0.4914 0.4144 0.7305
SPREAT 0.7416 0.5637 0.4704 0.5594 0.4460 0.6613
CTM 0.7219 0.5595 0.4930 0.5268 0.4530 0.6613
SOAP+c2m 0.7061 0.5887 0.4817 0.5455 0.4599 0.6400
SPREAT+c2m 0.7298 0.5804 0.4592 0.5455 0.4739 0.6240
Baranwal, M., Magner, A., Elvati, P., Saldinger, J., Violi,
A., and Hero, A. O. (2020). A deep learning architec-
ture for metabolic pathway prediction. Bioinformat-
ics, 36(8):2547–2553.
Bien, J. and Tibshirani, R. J. (2011). Sparse estimation of a
covariance matrix. Biometrika, 98(4):807–820.
Blei, D. and Lafferty, J. (2006). Correlated topic models.
Advances in neural information processing systems,
18:147.
Blei, D. M., Ng, A. Y., and Jordan, M. I. (2003). Latent
Aggregating Statistically Correlated Metabolic Pathways Into Groups to Improve Prediction Performance
59

dirichlet allocation. Journal of machine Learning re-
search, 3(Jan):993–1022.
Caspi, R., Billington, R., Foerster, H., and et al.
(2016). Biocyc: Online resource for genome and
metabolic pathway analysis. The FASEB Journal, 30(1
Supplement):lb192–lb192.
Caspi, R., Billington, R., Keseler, I. M., Kothari, A., Krum-
menacker, M., Midford, P. E., Ong, W. K., Paley, S.,
Subhraveti, P., and Karp, P. D. (2019). The metacyc
database of metabolic pathways and enzymes-a 2019
update. Nucleic acids research.
Dale, J. M., Popescu, L., and Karp, P. D. (2010). Machine
learning methods for metabolic pathway prediction.
BMC bioinformatics, 11(1):1.
Fan, J., Liao, Y., and Liu, H. (2016). An overview of the
estimation of large covariance and precision matrices.
The Econometrics Journal, 19(1):C1–C32.
Hahn, A. S., Konwar, K. M., Louca, S., and et al. (2016).
The information science of microbial ecology. Cur-
rent opinion in microbiology, 31:209–216.
Hanson, N. W., Konwar, K. M., Hawley, A. K., and
et al.(2014). Metabolic pathways for the whole com-
munity. BMC genomics, 15(1):1.
Hassa, J., Maus, I., Off, S., P
¨
uhler, A., Scherer, P., Klocke,
M., and Schl
¨
uter, A. (2018). Metagenome, metatran-
scriptome, and metaproteome approaches unraveled
compositions and functional relationships of micro-
bial communities residing in biogas plants. Applied
microbiology and biotechnology, 102(12):5045–5063.
He, J., Hu, Z., Berg-Kirkpatrick, T., and et al. (2017). Effi-
cient correlated topic modeling with topic embedding.
In Proceedings of the 23rd ACM SIGKDD Interna-
tional Conference on Knowledge Discovery and Data
Mining, pages 225–233. ACM.
Hoffman, M. D., Blei, D. M., Wang, C., and Paisley,
J. W.(2013). Stochastic variational inference. Jour-
nal of Machine Learning Research, 14(1):1303–1347.
Jiao, D., Ye, Y., and Tang, H. (2013). Probabilistic infer-
ence of biochemical reactions in microbial communi-
ties from metagenomic sequences. PLoS Comput Biol,
9(3):e1002981.
Kanehisa, M., Furumichi, M., Tanabe, M., and et al.
(2017). Kegg: new perspectives on genomes, path-
ways, diseases and drugs. Nucleic Acids Research,
45(D1):D353–D361.
Karp, P. D., Latendresse, M., Paley, S. M., and et al. (2016).
Pathway tools version 19.0 update: software for path-
way/genome informatics and systems biology. Brief-
ings in bioinformatics, 17(5):877–890.
Khatri, P., Sirota, M., and Butte, A. J. (2012). Ten years
of pathway analysis: current approaches and out-
standing challenges. PLoS computational biology,
8(2):e1002375.
Lawson, C. E., Harcombe, W. R., Hatzenpichler, R., and
et al. (2019). Common principles and best practices
for engineering microbiomes. Nature Reviews Micro-
biology, pages 1–17.
Lin, T., Tian, W., Mei, Q., and Cheng, H.(2014). The
dual-sparse topic model: mining focused topics and
focused terms in short text. In Proceedings of the 23rd
international conference on World wide web, pages
539–550. ACM.
Loh, P.-R., Baym, M., and Berger, B. (2012). Compressive
genomics. Nature biotechnology, 30(7):627.
M. A. Basher, A. R. and Hallam, S. J. (2021). Relabeling
metabolic pathway data with groups to improve pre-
diction outcomes. BioRxiv.
M. A. Basher, A. R., McLaughlin, R. J., and Hallam, S. J.
(2020). Metabolic pathway inference using multi-
label classification with rich pathway features. PLOS
Computational Biology, 16(10):1–22.
M. A. Basher, A. R., McLaughlin, R. J., and Hallam, S. J.
(2021a). Metabolic pathway prediction using non-
negative matrix factorization with improved precision.
Journal of Computational Biology.
M. A. Basher, A. R., McLaughlin, R. J., and Hallam,
S. J. (2021b). Metabolic pathway prediction using
non-negative matrix factorization with improved pre-
cision. In Computational Advances in Bio and Med-
ical Sciences, pages 33–44, Cham. Springer Interna-
tional Publishing.
Mascher, M., Schreiber, M., Scholz, U., Graner, A.,
Reif, J. C., and Stein, N. (2019). Genebank ge-
nomics bridges the gap between the conservation of
crop diversity and plant breeding. Nature genetics,
51(7):1076–1081.
Qi, Q., Li, J., and Cheng, J. (2014). Reconstruction
of metabolic pathways by combining probabilistic
graphical model-based and knowledge-based meth-
ods. In BMC proceedings, volume 8, pages 1–10.
Springer.
Sczyrba, A., Hofmann, P., Belmann, P., and et al. (2017).
Critical assessment of metagenome interpretation—a
benchmark of metagenomics software. Nature meth-
ods, 14(11):1063.
Shafiei, M., Dunn, K. A., Chipman, H., Gu, H.,
and Bielawski, J. P. (2014). Biomenet: A
bayesian model for inference of metabolic divergence
among microbial communities. PLoS Comput Biol,
10(11):e1003918.
Stewart, F. J., Sharma, A. K., Bryant, J. A., and et al. (2011).
Community transcriptomics reveals universal patterns
of protein sequence conservation in natural microbial
communities. Genome biology, 12(3):R26.
Tabei, Y., Yamanishi, Y., and Kotera, M. (2016). Simul-
taneous prediction of enzyme orthologs from chem-
ical transformation patterns for de novo metabolic
pathway reconstruction. Bioinformatics, 32(12):i278–
i287.
Hallam Lab(2021a). leADS: https://github.com/ hallam-
lab/leADS.
Hallam Lab(2021b). reMap: https://github.com/ hallam-
lab/reMap.
Wang, W.-L., Xu, S.-Y., Ren, Z.-G., Tao, L., Jiang, J.-W.,
and Zheng, S.-S.(2015). Application of metagenomics
in the human gut microbiome. World journal of gas-
troenterology: WJG, 21(3):803.
Yamanishi, Y., Tabei, Y., and Kotera, M. (2015).
Metabolome-scale de novo pathway reconstruction
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
60

using regioisomer-sensitive graph alignments. Bioin-
formatics, 31(12):i161–i170.
Ye, Y. and Doak, T. G. (2009). A parsimony approach
to biological pathway reconstruction/inference for
genomes and metagenomes. PLoS Comput Biol,
5(8):e1000465.
Zhao, Y., Chen, M.-H., Pei, B., Rowe, D., Shin, D.-G.,
Xie, W., Yu, F., and Kuo, L. (2012). A bayesian ap-
proach to pathway analysis by integrating gene–gene
functional directions and microarray data. Statistics in
biosciences, 4(1):105–131.
Aggregating Statistically Correlated Metabolic Pathways Into Groups to Improve Prediction Performance
61
