
Analysis of Impact of European Medical Device Regulation and
Brexit on the Regulatory Approaches in a Clinical Investigation
Study on a New Class III Medical Devices Conducted in Europe and
United Kingdom
Candice Houg
1,2
a
, Thomas Lihoreau
1,2
b
, Martina Hennessy
3
c
, Helene Esperou
4
d
,
Rachel Benamore
5
e
, Jean Palussiere
6
f
and Lionel Pazart
1,2
g
1
Centre Hospitalier Universitaire de Besançon, Centre d'Investigation Clinique,
INSERM CIC 1431, 25030, Besançon, France
2
Tech4Health network - FCRIN, France
3
Wellcome-HRB Clinical Research Facility, St James’s Hospital and School of Medicine Trinity College Dublin,
Dublin, Ireland
4
Clinical Research Unit, National Institute of Health and Medical Research, Paris, France
5
Department of Radiology, Oxford University Hospital Trust, Oxford, U.K.
6
Department of Interventional Radiology, Bergonie Institute, Bordeaux, France
Keywords: Clinical Investigation, Regulatory Approach, Medical Device, CE Marking, European Medical Device
Regulation 2017/745, Brexit, Ethics Committee, Competent Authority, High-risk Device.
Abstract: The evolution of technological innovations and medical devices requires particular reflections in terms of
regulation. In order to harmonise practices between European countries and to reinforce clinical investigations,
the European Regulation on medical devices 2017/745 has come to give a regulatory framework to the world
of devices. A summary of the regulatory approaches for a clinical investigation of a new class III device
conducted in France, Ireland and England is proposed in this article to illustrate the complexity of the
processes, ending with an example. This illustrates the impact of the EU regulation and Brexit on the conduct
of clinical investigations.
1 INTRODUCTION
Clinical investigations conducted in Europe were
regulated until the end of May 2021 by the Directive
90/385/EEC on active implantable medical devices
(EUR-lex, 1990) and the Directive 93/42/EEC on
medical devices (EUR-lex, 1993). Each European
country could thus transpose the directives into its
national law, as for example France with the Jardé
law (Legifrance, 2016), which separated research into
three categories according to the risks incurred for the
persons participating in this research.
a
https://orcid.org/0000-0002-8028-276X
b
https://orcid.org/0000-0001-8417-6609
c
https://orcid.org/0000-0002-2153-5288
d
https://orcid.org/0000-0002-1654-4868
e
https://orcid.org/0000-0001-7186-2677
f
https://orcid.org/0000-0001-6118-8543
g
https://orcid.org/0000-0002-9104-0862
The year 2021 is a year of major regulatory
change, including the European medical device
regulation and Brexit. The implementation of the
European regulation on medical devices 2017/745
(EUR-lex, 2017) has thus aimed to harmonise
practices between European countries. During 2021,
the United Kingdom (UK) separates from the
European Union (EU), known as Brexit, so that all
European laws and regulations no longer apply in the
UK, including the new European Medical Device
Regulation.
250
Houg, C., Lihoreau, T., Hennessy, M., Esperou, H., Benamore, R., Palussiere, J. and Pazart, L.
Analysis of Impact of European Medical Device Regulation and Brexit on the Regulatory Approaches in a Clinical Investigation Study on a New Class III Medical Devices Conducted in Europe
and United Kingdom.
DOI: 10.5220/0010968600003123
In Proceedings of the 15th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2022) - Volume 1: BIODEVICES, pages 250-258
ISBN: 978-989-758-552-4; ISSN: 2184-4305
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

Here, we present the impact of these changes on
the preparation and regulatory submission of a
clinical investigation of a new class III medical
device.
2 MEDICAL DEVICE
REGULATION 2017/745
The European Medical Device Regulation 2017/745
(EU MDR) entered into force on 26 May 2021 after a
year of delay due to the Covid crisis (EUR-lex, 2017).
The EU MDR replaces Directives 90/385/EEC and
93/42/EEC on active implantable medical devices
and medical devices respectively. A regulation,
unlike a directive, is not transposed into the national
regulations of each country; the Member States must
apply it in full and directly. The regulation therefore
aims to harmonise practices within Europe.
This EU MDR aims to strengthen market
surveillance and the clinical evaluation process,
to improve transparency through the European
Database on Medical Devices (Eudamed)
(https://ec.europa.eu/tools/eudamed/) and the unique
device identifier (UDI), and to strengthen the quality
and missions of notified bodies.
Manufacturers in order to market their device
must obtain the CE marking. CE marking (figure1) is
a guarantee that the product meets the essential safety
and performance European requirements.
Figure 1: CE marking.
The manufacturer must therefore provide
evidence of conformity with the requirements in
accordance with Article 5 of the EU MDR: "The
demonstration of conformity with the general safety
and performance requirements shall include a clinical
evaluation as provided for in Article 61".
The purpose of this clinical evaluation is to collect
clinical data on the medical device in order to verify,
under normal conditions of use, that its performance
corresponds to that claimed, to identify any
undesirable side effects and to assess the risks for the
patient.
Manufacturers, in order to demonstrate
compliance with the essential requirements, must
plan, perform and document a clinical evaluation of
the medical device. The clinical evaluation may be
based on:
- a critical evaluation of scientific publications
on equivalent devices
- a critical evaluation of the results of clinical
investigations
- and the consideration of currently available
alternatives
EU MDR specifies that, in the case of implantable
devices and Class III devices, clinical investigations
(CI) must be conducted. However, manufacturers are
not required to conduct a clinical investigation if the
following three criteria are met:
- the device has been designed by modifying a
device already marketed by the same
manufacturer
- equivalence with that device is demonstrated
and approved by a Notified Body
- the clinical evaluation of the device currently
marketed is sufficient to demonstrate
compliance of the modified device with the
relevant safety and performance requirements.
In the case of a brand new class III product
without equivalent on the EU market, clinical
investigation is required for its marketing in all
European countries.
3 BREXIT AND CLINICAL
INVESTIGATION
The United Kingdom (England, Scotland, Wales and
Northern Ireland) withdrew from the EU on 1
February 2020. The withdrawal agreement (EUR-lex,
2019) between the EU and the UK provided for a
transition period until 31 December 2020. The UK is
thus considered a "third country" by the EU as of 1
January 2021.
3.1 Regulation of Medical Device and
Clinical Investigation
Although the Medical Devices Regulation 2017/745
was written and came into force in 2017 when the UK
was still part of the European Union, medical devices
and clinical investigations in UK are not covered by
this EU regulation anymore. Medical devices remain
regulated by the UK Medical Device Regulation 2002
(Legislation.gov.uk, 2002). This regulation is the
adaptation of the European Directive 90/385/EEC
and the European Directive 93/42/EEC into UK law.
Analysis of Impact of European Medical Device Regulation and Brexit on the Regulatory Approaches in a Clinical Investigation Study on a
New Class III Medical Devices Conducted in Europe and United Kingdom
251

3.2 The CE Mark and UKCA Mark in
UK
Following the UK's separation from the European
Union, the CE mark, which guarantees the conformity
of devices to the essential performance and safety
requirements of the EU, is no longer applicable to
medical devices in the UK. Medical devices must
now be UKCA (UK Conformity Assessed) certified
in order to move freely in the UK. There is a transition
period for CE marked devices to be recognised in the
UK until 1
st
January 2023, allowing manufacturers to
build up the required dossiers for UKCA marking
(figure 2). The UKCA marking is, in some ways,
similar to the CE marking as the majority of UK
standards follow the European standards. One of the
main differences lies in the bodies responsible for
issuing the UK or CE mark.
Figure 2: UKCA marking.
The UKCA certificate must be issued by
approved Notified Bodies responsible for assessing
the conformity of the device with UK requirements.
The guide "UK approved bodies for medical
devices" listing the notified bodies is available on
the UK government website (https://www.gov.uk/
government/publications/medical-devices-uk-
approved-bodies/).
UK-based notified bodies, which were competent
to assess the conformity of European products and
issue the CE certificate, are no longer recognised in
the EU and therefore can no longer issue these CE
mark certificates.
3.3 Data Protection
In the course of a clinical investigation, the personal
and health data of participants are processed,
collected and analysed for scientific research
purposes. The sponsor, person or institution
responsible for the implementation, management or
financing of a clinical study is thus responsible for the
protection and confidentiality of collection, transfer
and treatment of personal data during the study.
The General Data Protection Regulation (GDPR)
(EUR-lex, 2016) governs the processing of personal
data and the rules on the free movement of personal
data in Europe. The UK has special provisions
regarding the GDPR. Thanks to the Trade and
Cooperation Agreement (EUR-lex, 2021b) concluded
on 24
th
December 2020, the GDPR remained in force
throughout the UK until 1
st
July 2021. This meant that
data transfer with the UK could take place under the
terms of the GDPR until 1
st
July 2021 without it being
considered a third country. After 1
st
July, if there was
no European Commission decision authorising the
transfer of personal data to the UK ("adequacy
decision"), the country would have been listed as a
third country for the transfer of data and the UK
would have had to demonstrate that it had a sufficient
and adequate level of data protection for transfers to
continue. Instead, the European Commission adopted
an adequacy decision on the UK and the General Data
Protection Regulation on 28
th
June 2021 (EUR-lex,
2021a). The European Commission found, through its
decisions, that the UK enjoys a level of protection
substantially equivalent to that guaranteed by EU law
and thus transfers of personal data from the EU to the
UK could proceed without further specific directives.
4 REGULATORY APPROACHES
FOR CLINICAL
INVESTIGATION
In the EU MDR, clinical investigation is defined as
“any systematic investigation involving one or more
human participants to assess the safety or
performance of a device”. Clinical investigations are
time-consuming and expensive studies with complex
regulatory procedures.
4.1 Multinational Clinical Investigation
Multinational clinical investigations are
investigations conducted with a common
methodology in more than one country and a common
recruitment pool across all participating countries. In
this way, the multinational dimension allows access
to a larger number of subjects and thus reduces the
duration of the study and its cost while also improving
generalisability of participant characteristics. It
allows the device to be evaluated in different
environments and so ensures that it is compatible with
different organisations.
Finally, the results can be extrapolated more
easily as the study is conducted in more
ClinMed 2022 - Special Session on Dealing with the Change in European Regulations for Medical Devices
252

representative country and manufacturers benefit
from better exposure of their product, which can
facilitate its market penetration once it has been CE
marked.
The main challenge of multinational studies is to
apply for a clinical investigation authorisation from
the regulatory authorities - ethics committee and
competent authority. Although European projects are
subject to the same regulations, these regulations
leave some room for manoeuvre to national law.
Moreover, evaluation of the study by the ethics
committee is specific to each country and the
procedures for submission to the competent
authorities of each Member State are not harmonised
between European countries as yet and until
establishment of this function under the Eudamed
platform.
4.2 Common Rules for Clinical
Investigations
Clinical investigation, regardless of their size
(monocentric, multicentre, international) and their
purpose (compliance with essential requirements,
post-marketing clinical follow-up, etc.), must be
conducted in accordance with rules on ethics and
good clinical practice.
4.2.1 Ethics Rules
A clinical investigation must be designed and
conducted in an ethical manner. The first ethics
principles were proclaimed in 1947 by the Nuremberg
Code, which followed the crimes against humanity
committed during the Second World War. This text
was then completed by the Helsinki Declaration
(Wold Medical Association, 2013) in 1964. These
international texts now constitute the key principles
of ethical research.
4.2.2 Conduct Rules
The clinical investigation should also be designed and
conducted in accordance with good clinical practice.
ICH Good Clinical Practice (GCP: ICH E6(R2)) is an
international ethical and scientific quality standard
for clinical trials involving human subjects
(International Council for Harmonisation of
Technical Requirements for Pharmaceuticals for
Human Use, 2016). This standard has its origin in the
Declaration of Helsinki. GCP: ICH E6 describes a
standard for the design, conduct, recording and
reporting of clinical studies.
Recently, an expert group has established the ISO
14155 (International Organization for
Standardization, 2020) standard which cites Good
Clinical Practice specific to clinical investigations.
This standard is based on ICH GCP E6: R2 and uses
terminology more appropriate to medical devices.
ISO 14155 has its origins in the Declaration of
Helsinki, whose objective is to protect the rights,
safety and well-being of subjects and to ensure that
these principles prevail over the interests of science
and society.
The ISO 14155 standard states in part that a
clinical investigation:
- must be conducted under the responsibility of
a sponsor and should be conducted at the
research site by qualified investigators
- must be conducted in accordance with a
clinical investigation plan (protocol)
- must have received the approval/favourable
opinion of the local ethics committee
- must have received no objection from the local
regulatory authorities (if applicable)
- the subject must have been adequately
informed about their participation and the
risks involved. The subject must have freely
given consent before participating in the
clinical investigation
- medical devices used in clinical investigations
should be used in accordance with the
investigator's brochure, the protocol and the
instructions for use
4.2.3 Data Protection Rules
Clinical investigations conducted in the EU and
United Kingdom must also comply with the GDPR.
To ensure the protection of the rights and freedoms of
individuals, technical and organisational measures
must be taken.
The study sponsor must therefore ensure that the
study complies with the GDPR, since health data,
which is both personal data and sensitive data, is
processed. The person must be informed about the
processing of his data and give his consent to the
processing of his data.
4.3 Regulation of Clinical Investigation
in Europe
Clinical investigations are governed in Europe by the
European Medical Devices Regulation 2017/745. The
establishment of a common regulation as EU MDR is
a real opportunity to harmonize the evaluation time
but also to develop a system of vigilance of medical
Analysis of Impact of European Medical Device Regulation and Brexit on the Regulatory Approaches in a Clinical Investigation Study on a
New Class III Medical Devices Conducted in Europe and United Kingdom
253

devices, absent until now. Chapter VI of the EU MDR
is entirely devoted to the regulation of clinical
evaluations and clinical investigations.
For medical devices of class III, invasive and
implantable device, the study must get the following
requirements:
- validated by the Member State: it must ensure
that the study falls within the scope of the EU
MDR and that the application dossier is
complete
- authorised by the Member State after a full
assessment of the application file
- authorised by the Ethics Committee after
evaluation of the dossier
- covered by insurance/indemnity in case of
injury to participants due to their participation
in the research
- any adverse events must be recorded and
reported to the Member State
- carried out under the responsibility of a
sponsor established in the UE
For clinical investigations of class I medical
devices and class IIa and IIb non-invasive devices, the
requirements are the same, except that the study does
not require Member State authorisation. Article 70
paragraph 7a stipulates that clinical studies require
only a validation from the Member state and the
favourable opinion of the ethics committee unless
otherwise stated by national law. French and Irish law
make use of the opening clause in Article 70(7a) for
clinical investigations with low risk medical devices.
Annex XV of MDR details documents to submit
to member states for validation and/or authorisation
by the member state. Each Member State may request
the submission of specific documents to make its
assessment. Files to submit to the ethics committee
are dependent on the local committee.
The EU MDR specifies that the application for a
clinical investigation must be submitted via the
Eudamed and that the summary and results of the
application must be filed on the portal. The clinical
investigation module of the Eudamed electronic
system is currently not available and will only be
deployed from 2022 onwards, the application must be
made according to national procedures during the
transitional period according to the MDCG 2021-16
(Medical Devices Coordination Group, 2020).
4.3.1 Specific Country Regulation
We will present the specific regulation in three
countries by detailing the regulatory authority with
responsibility for the clinical investigation, the
documents required for submission and the
evaluation timeframes: France, Ireland and England
France. The competent authority in France is the
National Agency for Medicines and Health Products
– ANSM (https://ansm.sante.fr/). It is in charge of
authorising and monitoring clinical studies on
medicines, medical devices, non-health products and
cosmetics in France.
Ethics committee responsible for issuing an
opinion on research projects in France is the “Comité
de Protection des Personnes (CPP)”. There are 40
CPPs in France and the opinion of a single CPP is
required at national level, regardless of the number of
centres. The study files will be submitted on a
national platform and the appointment of a CPP is
done by drawing lots.
The requirements specific to the class of the
device are summarised in the Table 1 and the
difference from the EU MDR are indicated by (*),
using of the opening clause in Article 70(7a).
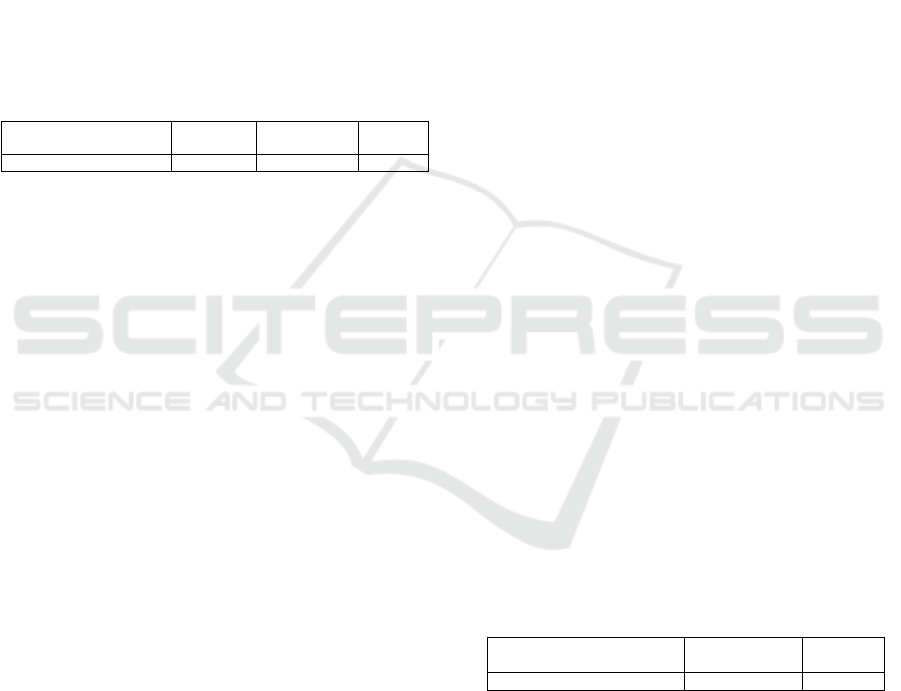
Table 1: Regulatory procedures required in France to
conduct a clinical investigation to establish the conformity
of a medical device according to its class.
ANSM
validation
ANSM
authorisation
CPP
o
p
inion
Class I
Class IIa non-invasive
x x
Class IIb non-invasive*
Class IIa and IIb invasive
Class III
x x x
Researchers and manufacturers can refer to the
guide "Avis aux promoteurs - Investigations cliniques
de dispositifs médicaux relevant du Réglement
Européen N° 2017/745 Partie I" (ANSM, 2021)
available on the ANSM website for the conduct of
their clinical investigations.
To obtain authorisations from the regulatory
authorities, the applicant must first obtain an IDRCB
registration number for its research and obtain the
designation of a CPP. The complete application file
must then be submitted to the designated CPP and to
the ANSM on the same day but separately: by email
or on the Eudralink platform for the ANSM
(https://eudralink.ema.europa.eu/) and on the
CNRIPH platform for the CPP
(https://cnriph.sante.gouv.fr/).
The ANSM has 10 days to validate the application
in accordance with the EU MDR. Then the ANSM
and the CPP must give their opinion within 45 days.
This period may be extended by 20 days by the
ANSM if experts’ consultation is needed.
ClinMed 2022 - Special Session on Dealing with the Change in European Regulations for Medical Devices
254
Ireland. The Health Products Regulatory Authority –
HPRA - (http://www.hpra.ie/homepage/medical-
devices) is the regulatory authority for health
products as medicines, medical devices, cosmetics for
humans and animals in Ireland. The HPRA is the
authority responsible for assessing and authorising
clinical trials of medicines and medical devices.
The ethics committee in Ireland is the National
Research Ethics Committee - NREC, a national ethics
committee (https://www.nrecoffice.ie/).
The requirements specific to the class of the
device are summarised in the Table 2 and the
difference from the EU MDR are indicated by (*),
using of the opening clause in Article 70(7a).
Table 2: Regulatory procedures required in Ireland to
conduct a clinical investigation to establish the conformity
of a medical device according to its class.
HPRA
validation
HPRA
authorisation
NREC
o
p
inion
All class of devices* * x x
The "Guide to Clinical Investigations Carried Out
in Ireland"(HPRA, 2021a) is a reference for
researchers and manufacturers.
The authorisation application to be submitted to
the HPRA must be filed on the Common European
Submission Portal (https://cespportal.hma.eu/). For
the ethics committee, the file must be sent by email to
the NREC.
The HPRA has a period of 45 calendar days to
evaluate the application after validation of the file.
The HPRA may consult experts and an additional 20
calendar days is added. The NREC meets once a
month to assess applications. The investigator must
submit the application 12 days before the date of the
plenary meeting and receives a response within 55
days of meeting.
The sponsor must pay fees to the HPRA (HPRA,
2021b) and NREC (https://www.nrecoffice.ie/apply-
2/fees/) for their initial evaluation of the application.
NREC fees are function of industrial or academic lead
and raised at 500€ and 75€ respectively. Fees for
HPRA are dependant of the class of the device:
- class III and IIb medical devices or active
implantable device: 4300€
- class IIa and class I medical devices: 1900€
In case of substantial amendment or resubmission,
supplementary fees are required.
4.3.2 Regulation of Clinical Investigation in
UK
The United Kingdom is a "third country" by the EU.
The EU Medical Devices Regulation 2017/745
therefore does not apply to the UK and clinical
investigations in the UK are governed by the UK
Medical Devices Regulations 2002
(Legislation.gov.uk, 2002).
For a clinical investigation of all class of devices,
the following requirements are needed:
- sponsor is established in UK or in a country
listed in the EU and/or the European
Economic Area.
- a favourable opinion from the ethics
committee
- the authorisation of the competent authority
- the consent of each included subjects
- an insurance/indemnity in case of injury
- to report the adverse event
The competent authority in United Kingdom is
the Medicines and Healthcare Products Regulatory
Agency – MHRA (https://www.gov.uk/government/
organisations/medicines-and-healthcare-products-
regulatory-agency). The MHRA is the body
responsible for assessing and authorising applications
for clinical investigations.
There are over 80 different Research Ethics
Committees (RECs) in the UK within the Research
Ethics Department of the UK Departments of Health
(https://www.hra.nhs.uk/about-us/committees-and-
services/res-and-recs/search-research-ethics-
committees/). RECs are classified as "flagged RECs"
according to the professional, academic and ethical
expertise of the committee members. For clinical
investigations, 10 flagged RECs are listed in England.
Approval of only one REC is required, regardless of
the number of centres involved in the clinical
investigation.
The requirements specific to the class of the
device are summarised in the Table 3.
Table 3: Regulatory procedures required in England to
conduct a clinical investigation to establish the conformity
of a medical device according to its class.
MHRA
authorisation
REC
opinion
All class of devices x x
The application to the MHRA and the REC must
be submitted on the Integrated Research Application
System by the principal investigator of the research in
England (https://www.myresearchproject.org.uk/).
Where the clinical investigation involves the NHS,
patients or NHS staff, approval from the HRA is
required. This application is made in conjunction with
the REC application. The MHRA and the REC each
have 5 days to confirm receipt and completeness of
the application after receipt and 60 days to assess the
application.
Analysis of Impact of European Medical Device Regulation and Brexit on the Regulatory Approaches in a Clinical Investigation Study on a
New Class III Medical Devices Conducted in Europe and United Kingdom
255

The sponsor must pay a fee for the initial
evaluation of the application by the MHRA
(https://www.gov.uk/government/publications/mhra-
fees/current-mhra-fees), which depends on the class
of the device:
- Group A includes Class I, IIa and IIb devices
other than long-term implantable/invasive
devices: £3820
- Group B includes Class IIb implantable/long
term invasive, Class III, active implantable
devices: £5040
In case of amendment or resubmission,
supplementary fees are required by MHRA.
5 PRACTICAL EXAMPLE
The Selio project is a multi-partner European funded,
project on the development of a new medical device
which will be class III in Europe. This project is
supported by EIT Health. It was born during the
period of transition from the European Medical
Device Directive to the European Medical Device
Regulation and the separation of the UK from the
European Union. The EU Medical Device Regulation
2017/745 and Brexit has directly affected the project
and the regulatory steps required to obtain
authorisations to start a clinical investigation
involving French, Irish and English partners. We
present here the expected flow chart of regulatory
steps for the preparation and submission of a clinical
investigation on a new class III medical device.
Class III medical device products without
equivalent on the EU market, require a clinical
investigation in the framework of the clinical
evaluation. A Notified Body will then have to assess
the conformity of the device with the European
requirements in terms of safety and performance in
order to issue the CE mark.
In order to reduce recruitment time and increase
recognition of scientific value, one option for this
project is to conduct a multinational clinical study
with French, Irish and English centres. Science is
stronger when it is collaborative. The UK has been
one of the most important scientific partners in
Europe for decades. Their lack of participation in
such large-scale projects, due to policies different
from those of Europe or regulatory procedures too
complex to include them in such projects, would have
an impact on the value of science.
For the conduct of this clinical investigation, the
sponsor will have to prepare and submit an
application for authorisation in each of the countries
participating in the study. He will thus have to prepare
the documents required for the competent authorities
and ethics committees for their evaluation of the
study and their authorisation.
Although some of the documents are common -
the clinical investigation plan, the information note
and consent form, the investigator's brochure and the
proof of insurance - the latter part of the documents is
specific to each authority and thus requires additional
time and regulatory expertise to draft.
The work required for a multinational study is
much more time and resource intensive than a
national study. It is necessary to have a regulatory
contact in each of the countries participating in the
study for the preparation of the regulatory procedures.
The project team is composed of scientific experts,
project managers, clinical research associates and
clinical study technicians. In addition, an operational
and scientific committee participates in the
construction, validation and follow-up of the study.
The study can start in a given country once the
competent authority has given its authorisation and
the ethics committee has given a favourable opinion.
In an ideal situation, which means without the need
for the regulatory authorities to consult experts or
issue comments and/or modifications to the research,
the research could start approximately two months
after the submission of the application in each country
(table 4).
Table 4: Delay of assessment in days for competent
authorities and ethics committee of France, Ireland and
England. The delay indicated are minimal delay, i.e.
without expert consultation or question to the sponsor.
Competent authority Ethics
committee
France 55 55
Ireland 45 + x days for
validation
55
England 65 65
In most cases, studies receives from regulatory
authorities opinions subject to minor or major
changes, which can extend the assessment period by
up to 6 months. Furthermore, in the case of
multinational studies, requests for changes to the
study protocol must be carried over all countries in
the form of amendments. This not only lengthens the
evaluation periods but also leads to additional costs
when the activities of the regulatory authorities are
invoiced.
ClinMed 2022 - Special Session on Dealing with the Change in European Regulations for Medical Devices
256

In accordance with the previously announced
costs, the budget for the initial submission of a
clinical study to the regulatory authorities can amount
to more than 10,000€. The budget is consequent and
should not be neglected during the financial set-up.
The variation in evaluation time and cost is
significant when considering best and worst case
scenarios. Investigators and project leaders need to be
able to explain this to funders and investors.
6 WHAT ABOUT THE
COORDINATED EVALUATION
PROCEDURE WITH THE EU
MDR AND THE EUDAMED
PLATFORM?
A coordinated evaluation procedure for clinical
investigations taking place in more than one Member
State will be introduced with the establishment of the
Eudamed, and this procedure will be made mandatory
for European clinical investigations from 26 May
2027.
The coordinated evaluation procedure will thus
simplify the sponsor's procedures, who will only have
to submit one application for authorisation of a clinical
investigation in Europe, regardless of the number of
European countries participating in the study.
A coordinating Member State will be identified
among the Member States participating in the clinical
investigation. The coordinating Member State will be
responsible for assessing whether the clinical
investigation falls within the scope of the EU MDR,
for verifying that the application is complete in
accordance with Annex XV with the exception of
certain documents which are subject to assessment by
each Member State, and for issuing an assessment
report. This report will have to be communicated to
the Member States in order to obtain their comments
on the project. The coordinating Member State will
then have to issue a final evaluation report to the
sponsor taking into account the comments of the
Member States within 45 days of the validation of the
application. This period may be extended to 95 days
if the study concerns a class IIb or III DM and expert
consultation is required.
7 CONCLUSION
Clinical investigations are long and costly studies.
The regulatory approaches could be cumbersome,
even more in the context of a multinational study. The
submission system to be used differ from one country
to another, the documents expected by the regulatory
authorities must be adapted to each regulatory
authority in respect of their national regulations and
the time period for the evaluation is also different and
dependant of certain factors. One potential risk could
be that investigators would seek to limit their study to
one jurisdiction or leave the UK out, resulting in poor
science and less confidence in the quality and
applicability of the devices after authorisation.
Although the European regulation tends to
harmonise practices for European countries, in the
absence of the Eudamed platform, these remain
complex and can therefore be a hindrance to
conducting a study of this scale.
The main advice to be drawn from this example is
that the sponsor should surround himself with people
with appropriate regulatory expertise at the design
stage of the project. For example identifying a
regulatory contact in each of the countries involved in
the study, which will enable him to be informed of the
regulatory steps to be taken for his clinical
investigation: the documents to be prepared, the
authorities in charge of the evaluation and the
deadlines to be respected.
The time needed for the assessment of the study
by the regulatory authorities should not be neglected
when planning the study (best or worst case scenario).
This time, together with the preparation of regulatory
documents, can sometimes exceed one year,
especially in projects of this size. It should therefore
be anticipated for smart project management.
In Europe, project sponsors of multinational
clinical trials can also be supported by the European
Clinical Research Infrastructure Network (ECRIN),
which can conduct by delegation some sponsor tasks
(https://ecrin.org/).
ACKNOWLEDGMENTS
The Selio project has received funding from the EIT
Health (https://eithealth.eu/): ID 20186.
REFERENCES
ANSM. (2021). Avis aux promoteurs Investigations
cliniques de dispositifs médicaux relevant du
Réglement Européen N° 2017/745 Partie I.
EUR-lex. (1990). Council directive of 20 June 1990 on the
approximation of the laws of the Member States
relating to active implantable medical devices
Analysis of Impact of European Medical Device Regulation and Brexit on the Regulatory Approaches in a Clinical Investigation Study on a
New Class III Medical Devices Conducted in Europe and United Kingdom
257

(90/385/EEC). https://eur-lex.europa.eu/legal-content/
EN/TXT/PDF/?uri=CELEX:31990L0385&from=EN
EUR-lex. (1993). Council directive 93/42/EEC of 14 June
1993 concerning medical devices. Official Journal L
169 , 12/07/1993 P. 0001 - 0043; Finnish Special
Edition: Chapter 13 Volume 24 P. 0085 ; Swedish
Special Edition: Chapter 13 Volume 24 P. 0085 ;
https://eur-lex.europa.eu/legal-content/EN/TXT/HT
ML/?uri=CELEX:31993L0042&from=FR
EUR-lex. (2016). Regulation (EU) 2016/679 of the
European Parliament and of the Council of 27 April
2016 on the protection of natural persons with regard to
the processing of personal data and on the free
movement of such data, and repealing Directive
95/46/EC (General Data Protection Regulation).
Official Journal of the European Union. https://eur-
lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CE
LEX:32016R0679&from=FR
EUR-lex. (2017). Regulation (EU) 2017/745 of the
European Parliament and of the Council of 5 April 2017
on medical devices, amending Directive 2001/83/EC,
Regulation (EC) No 178/2002 and Regulation (EC) No
1223/2009 and repealing Council Directives
90/385/EEC and 93/42/EEC (Text with EEA relevance.
). http://data.europa.eu/eli/reg/2017/745/oj/eng
EUR-lex. (2019). Agreement on the withdrawal of the
United Kingdom of Great Britain and Northern Ireland
from the European Union and the European Atomic
Energy Community (2019/C 384 I/01). Official Journal
of the European Union. https://eur-lex.europa.eu/legal-
content/EN/TXT/?uri=CELEX%3A12019W%2FTXT
%2802%29
EUR-lex. (2021a). Commission Implementing Decision
(EU) 2021/915 of 4 June 2021 on standard contractual
clauses between controllers and processors under
Article 28(7) of Regulation (EU) 2016/679 of the
European Parliament and of the Council and Article
29(7) of Regulation (EU) 2018/1725 of the European
Parliament and of the Council (Text with EEA
relevance). http://data.europa.eu/eli/dec_impl/
2021/915/oj/eng
EUR-lex. (2021b). Trade and Cooperation Agreement
between the European Union and the European Atomic
Energy Community, of the one part, and the United
Kingdom of Great Britain and Northern Ireland, of the
other part. Official Journal of the European Union.
https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=
uriserv%3AOJ.L_.2021.149.01.0010.01.ENG&toc=OJ
%3AL%3A2021%3A149%3ATOC
HPRA. (2021a). Guide to Clinical Investigations Carried
Out in Ireland.
HPRA. (2021b). Guide to Fees for Human Products.
http://www.hpra.ie/docs/default-source/publications-
forms/guidance-documents/fin-g0002-guide-to-fees-
for-human-products-v27.pdf?sfvrsn=69
International Council for Harmonisation of Technical
Requirements for Pharmaceuticals for Human Use.
(2016). Guideline for good clinical practice E6(R2).
European Medicines Agency. https://www.ema.
europa.eu/en/documents/scientific-guideline/ich-e-6-
r2-guideline-good-clinical-practice-step-5_en.pdf
International Organization for Standardization. (2020). ISO
14155:2011 Clinical investigation of medical devices
for human subjects—Good clinical practice.
https://www.iso.org/cms/render/live/en/sites/isoorg/co
ntents/data/standard/04/55/45557.html
Legifrance. (2016). Article L1121-1—Code de la santé
publique. Légifrance. https://www.legifrance.gouv.fr/
codes/article_lc/LEGIARTI000032722870/
Legislation.gov.uk. (2002). The Medical Devices
Regulations 2002 (SI 2002 No 618). https://www.legis
lation.gov.uk/uksi/2002/618/introduction/made
Medical Devices Coordination Group. (2020). MDCG
2020-16 Guidance on Classification Rules for in vitro
Diagnostic Medical Devices under Regulation (EU)
2017/746 November 2020. https://ec.europa.eu/health/
sites/default/files/md_sector/docs/md_mdcg_2020_gui
dance_classification_ivd-md_en.pdf
Wold Medical Association. (2013). WMA - The World
Medical Association-WMA Declaration of Helsinki –
Ethical Principles for Medical Research Involving
Human Subjects. https://www.wma.net/policies-
post/wma-declaration-of-helsinki-ethical-principles-
for-medical-research-involving-human-subjects/
ClinMed 2022 - Special Session on Dealing with the Change in European Regulations for Medical Devices
258
