
End-to-End Multi-channel Neural Networks for Predicting Influenza a
Virus Hosts and Antigenic Types
Yanhua Xu
a
and Dominik Wojtczak
b
Department of Computer Science, University of Liverpool, U.K.
Keywords:
Machine Learning, Influenza Virus, Long Short-term Network, Convolutional Neural Network, Transformer.
Abstract:
Influenza occurs every season and occasionally causes pandemics. Despite its low mortality rate, influenza is a
major public health concern, as it can be complicated by severe diseases like pneumonia. A accurate and low-
cost method to predict the origin host and subtype of influenza viruses could help reduce virus transmission and
benefit resource-poor areas. In this work, we propose multi-channel neural networks to predict antigenic types
and hosts of influenza A viruses with hemagglutinin and neuraminidase protein sequences. An integrated data
set containing complete protein sequences were used to produce a pre-trained model, and two other data sets
were used for testing the model’s performance. One test set contained complete protein sequences, and another
test set contained incomplete protein sequences. The results suggest that multi-channel neural networks are
applicable and promising for predicting influenza A virus hosts and antigenic subtypes with complete and
partial protein sequences.
1 INTRODUCTION
Influenza is a highly contagious respiratory illness
that results in as many as 650,000 respiratory deaths
globally per year (Iuliano et al., 2018). Influenza
spreads mainly through droplets, aerosols, or by di-
rect contact (Lau et al., 2010), and up to 50% of infec-
tions are asymptomatic (Wilde et al., 1999). Influenza
can complicate a range of clinical problems associ-
ated with high fatality rates, including secondary bac-
terial pneumonia, primary viral pneumonia, chronic
kidney disease, acute renal failure, and heart fail-
ure (Watanabe, 2013), (Casas-Aparicio et al., 2018),
(England, 2020).
The influenza virus genome comprises several
segments of single-stranded ribonucleic acid (RNA).
The virus has four genera, differentiated mainly by
the antigenic properties of the nucleocapsid (NP) and
matrix (M) proteins (Shaw and Palese, 2013). At
present, Influenza virus has four types: influenza
A virus (IAV), influenza B virus (IBV), influenza
C virus (IVC) and influenza D virus (IVD). IAV is
widespread in a variety of species, causes the most
serious diseases, and is the most capable of unleash-
ing a pandemic, while the others are less virulent. IAV
a
https://orcid.org/0000-0003-1028-9023
b
https://orcid.org/0000-0001-5560-0546
could trigger major public health disruption by evolv-
ing for efficient human transmission, as it did, with
the ‘Spanish Flu’, during 1918–1919, which is esti-
mated to have killed 20 to 100 million people (Mills
et al., 2004).
IVA is further subtyped by the antigenic properties
of its two surface glycoproteins, hemagglutinin (HA)
and neuraminidase (NA). There are presently 18 HA
subtypes and 11 NA subtypes known (Asha and Ku-
mar, 2019), of which only H1, H2, H3 and N1, N2
spread among humans. The avian influenza viruses
(H5N1, H5N2, H5N8, H7N7, and H9N2) may spread
from birds to humans; this occurs rarely but can be
deadly: all avian influenza A viruses have the poten-
tial to cause pandemics.
The virus uses HA and NA to bind to its host cells
(James and Whitley, 2017). HA allows the virus to
recognise and attach to specific receptors on host ep-
ithelial cells. Upon entering the host cell, the virus
replicates and is released by NA, thence infecting
more cells. The immune system can be triggered to
attack viruses and destroy infected tissue throughout
the respiratory system, but death can result through
organ failure or secondary infections.
Viruses undergo continuous evolution. Point mu-
tations in the genes that encode the HA and NA can
render the virus able to escape the immune system.
Such change is described as antigenic drift and leads
40
Xu, Y. and Wojtczak, D.
End-to-End Multi-channel Neural Networks for Predicting Influenza a Virus Hosts and Antigenic Types.
DOI: 10.5220/0011526300003335
In Proceedings of the 14th International Joint Conference on Knowledge Discovery, Knowledge Engineering and Knowledge Management (IC3K 2022) - Volume 1: KDIR, pages 40-50
ISBN: 978-989-758-614-9; ISSN: 2184-3228
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

to seasonal influenza. The other change, the antigenic
shift, occurs more rarely and results in a major change
in the production of a new virus that cannot be com-
pletely handled by the existing immune response, and
may leads to the pandemics (Clayville, 2011).
In this paper, we propose multi-channel neural
networks (CNN, bidirectional long short-term mem-
ory, bidirectional gated recurrent unit and trans-
former) to predict the subtypes and hosts of IAV.
The models were trained on an integrated protein se-
quence data set collected prior to 2019 (named pre-19
set) and tested both on an integrated data set collected
from 2019 to 2021 (named post-19 set), and a data set
containing incomplete sequences. We use Basic Lo-
cal Alignment Search Tool (BLAST) as the baseline
model and all models yield better performance than
the baseline model, especially multi-channel BiGRU.
Tested on the post-19 set, this model reaches 94.73%
(94.58%, 94.87%), 99.86% (99.82%, 99.89%) and
99.81% (99.74%, 99.89%) F
1
score for hosts, HA
subtypes and NA subtypes prediction, respectively.
The performance on incomplete sequences reaches
approximately 81.36% (80.35%, 82.37%), 96.86%
(96.50%, 97.21%) and 98.18% (97.80%, 98.56%) F
1
score for hosts, HA subtypes and NA subtypes pre-
diction, respectively.
2 RELATED WORK
Rapid and accurate detection of IAV hosts and sub-
types can improve influenza surveillance and re-
duce spread. The traditional methods for virus
subtyping, such as nucleic acid-based tests (NATs),
are labour intensive and time-consuming (Vemula
et al., 2016). Therefore, various supervised machine
learning-based methods have been developed to pre-
dict the hosts or subtypes of influenza viruses, based
on convolutional neural network (CNN) (Clayville,
2011), (Fabija
´
nska and Grabowski, 2019), (Scarafoni
et al., 2019), support vector machines (SVM) (Ah-
san and Ebrahimi, 2018), (Xu et al., 2017), (Kincaid,
2018), decision trees (DT) (Ahsan and Ebrahimi,
2018), (Attaluri et al., 2009), random forests (RF)
(Kincaid, 2018), (Eng et al., 2014), (Kwon et al.,
2020), etc.
The protein sequence is of variable length and
needs to be encoded as a numerical vector. Pre-
vious studies have sought to do so using simple
one-hot encoding (Clayville, 2011), (Eng et al.,
2014), (Mock et al., 2021), pre-defined binary en-
coding schemes (Attaluri et al., 2010), pre-defined
ASCII codes (Fabija
´
nska and Grabowski, 2019),
Word2Vec (Xu et al., 2017), and physicochemical
features(Chrysostomou et al., 2021), (Sherif et al.,
2017), (Kwon et al., 2020), (Yin et al., 2020). One
of the drawbacks of using handcrafted feature sets or
physicochemical features is they do require feature
selection or extraction process before training. There-
fore, we applied word embedding to ask models to
learn the features from given training data, which is
more convenient, straightforward and light-weighted.
Most work focuses on three classes (i.e. avian, swine
and human) or a single class of hosts from a single
database. In contrast to previous work, we collected
data from multiple databases and focuses on more re-
fined classes.
Multi-channel neural network has been used in re-
lation extraction (Chen et al., 2020), emotion recogni-
tion (Yang et al., 2018), face detection (George et al.,
2019), entity alignment (Cao et al., 2019), haptic ma-
terial classification (Kerzel et al., 2017), etc. Few
studies use the multi-channel neural networks in in-
fectious diseases and therein we proposed three kinds
of multi-channel neural network architectures to pre-
dict influenza A virus host and subtypes simultane-
ously, instead of training single task-specific models.
3 MATERIALS AND METHODS
3.1 Data Preparation
3.1.1 Protein Sequences
The complete hemagglutinin (HA) and neuraminidase
(NA) sequences were collected from the Influenza
Research Database (IRD) (Squires et al., 2012) and
Global Initiative on Sharing Avian Influenza Data
(GISAID) (Shu and McCauley, 2017) (status 16th
August 2021). The originally retrieved data set con-
tains 157,119 HA sequences and 156,925 NA se-
quences from GISAID, 96,412 HA sequences and
84,186 NA sequences from IRD. The redundant and
multi-label sequences were filtered, and only one HA
sequence and one NA sequence for each strain were
included in the data set. Therefore, each strain has a
unique pair of HA and NA sequences and belongs to
one host and one subtype. Our data set is from differ-
ent sources, and we removed sequences from GISAID
if they were already in IRD before integration. Some
strains in GISAID belonging to H0N0, HA0 is an un-
cleaved protein that is not infectious, also have been
removed. The strains isolated prior to 2019 are used
to produce the pre-trained model and strains isolated
from 2019 to 2021 are used only for testing the per-
formance of models.
The incomplete HA and NA sequences were col-
End-to-End Multi-channel Neural Networks for Predicting Influenza a Virus Hosts and Antigenic Types
41

Table 1: Summary statistics of data sets.
Data Set (alias) # Total # IRD # GISAID
< 2019 (pre-19) 27, 884 26,704 1,108
2019 - 2021 (post-19) 2,716 2,206 510
Incomplete (incomplete) 8,325 8,325 /
lected from IRD (status 16th August 2021). The se-
quence is thought as complete if its length is the same
as the length of the actual genomic sequence (Shu
and McCauley, 2017). We download the database
and then filter the complete sequences to get incom-
plete sequences, as both complete and incomplete se-
quences form the Influenza database (all sequences =
complete sequences ∪ incomplete sequences). In-
complete sequences are only used for testing the per-
formance of models. The details of the data sets are
summarized in Table 1.
3.1.2 Label Reassignment
IRD and GISAID recorded 45 and 33 hosts, re-
spectively, of which only 6 are consistent in both
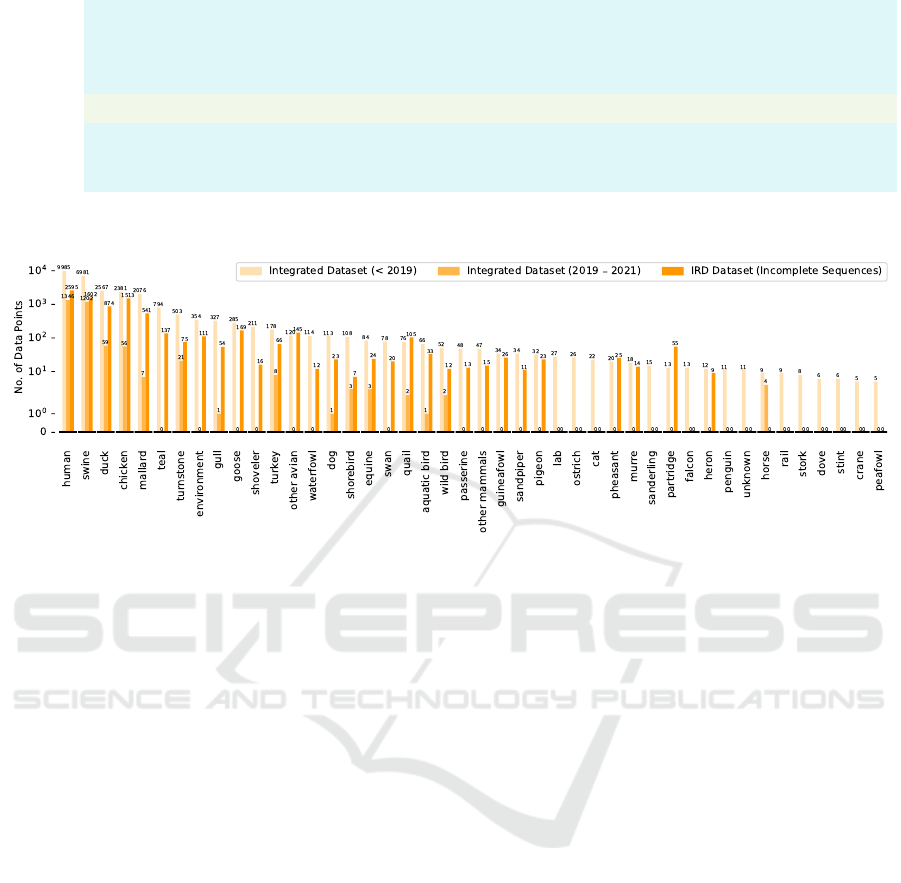
databases, as shown in Fig. 1. We regroup the host la-
bels into 44 categories, the distribution of regrouped
host labels is represented in Fig. 2. 18 HA (numbered
as H1 - H18) and 11 NA (numbered as N1 - N11) sub-
types have been discovered, respectively. We also re-
group very few subtypes in the data set into other sub-
types (i.e. H15, H17, H18, N10 and N11), as shown
in Fig. 3 and Fig. 4.
3.2 Sequence Representation
Neural networks are functional operators that perform
mathematical operations on inputs and generate nu-
merical outputs. A neural network cannot interpret
the raw sequences and needs them to be represented
as numerical vectors before feeding them to the neu-
ral network. The most intuitive and simple strategy
to vectorise the sequence is called one-hot encoding.
In natural language processing (NLP), the length of
the one-hot vector for each word is equal to the size
of vocabulary. The vocabulary consists of all unique
words (or tokens) in the data. If each amino acid is
represented as one “word”, then the length of the one-
hot vector for each amino acid depends on the num-
ber of unique amino acids. Therefore, one-hot encod-
ing results in a sparse matrix for large vocabularies,
which is very inefficient. A more powerful approach
is to represent each word as a distributed dense vector
by word embedding, which learns the word represen-
tation by looking at its surroundings, so that similar
words are given similar embeddings. Word embed-
ding has been successfully used to extract features of
biological sequences (Asgari and Mofrad, 2015).
A protein sequence can be represented as a set of
3-grams. In NLP, N-grams are N consecutive words
in the text, and N-grams of a protein sequence are N
consecutive amino acids. For example, the 3-grams
of sequence MENIVLLLAI is MEN ENI NIV IVL
VLL LLL LLA LAI. We set N as 3 as suggested by
previous research (Xu and Wojtczak, 2022).
3.3 Neural Network Architecture
We propose a multi-channel neural network architec-
ture that takes two inputs (HA trigrams and NA tri-
grams) and generates three outputs (host, HA sub-
types and NA subtypes). The neural networks applied
in this study include bidirectional long short-term
memory (BiLSTM), bidirectional gated recurrent unit
(BiGRU), convolutional neural network (CNN) and
Transformer.
3.3.1 Bidirectional Recurrent Neural Networks
We use two kinds of bidirectional recurrent network
networks in this study: Bidirectional Long Short-
Term Memory (BiLSTM) and Bidirectional Gated re-
current unit (BiGRU). LSTM is an extension of re-
current neural network (RNN). It uses gates to reg-
ulate the flow of information to tackle the vanish-
ing gradient problem of standard RNNs (Hochreiter
and Schmidhuber, 1997b), (Hochreiter and Schmid-
huber, 1997a). A common LSTM has three gates:
input gate, forget gate and output gate. The input
gate stores new information from the current input
and selectively updates the cell state, the forget gate
ignores irrelevant information, and the output gate de-
termines which information is moved to the next hid-
den state. Bidirectional LSTM (BiLSTM) (Graves
et al., 2005), (Thireou and Reczko, 2007) comprises
a forward LSTM and a backward LSTM to train the
data in both directions, leading to better context un-
derstanding, and is more effective than unidirectional
LSTM (Graves and Schmidhuber, 2005).
The Gated recurrent unit (GRU) is similar to
LSTM but only has a reset gate and an update gate
(Cho et al., 2014). The reset gate decides how much
previous information needs to be forgotten, and the
update gate decides how much information to discard
and how much new information to add. GPUs have
fewer tensor operations and are therefore faster than
LSTMs in terms of training speed. Bidirectional GRU
also includes forward and backward GRU.
KDIR 2022 - 14th International Conference on Knowledge Discovery and Information Retrieval
42
anteater, avian, bat, beetle, bovine, camel, caprine, civet, civet cat, crane, dog, domestic cat,
donkey, ferret, flat-faced bat, fowl, fox, horse, insect, large cat, lion, marten, meerkat, mink,
monkey, muskrat, panda, pika, plateau pika, raccoon, raccoon dog, rat, reassortant, sea mam-
mal, seal, skunk, weasel, wildebeest, yak
chicken, curlew, duck, eagle, falcon, goose, grouse, guinea fowl, gull, ostrich, other avian, par-
tridge, passerine, penguin, pheasant, pigeon, rails, sandpiper, shearwater, swan, turkey, turn-
stone, US quail, canine, equine, feline, other mammals
human, laboratory derived, unknown, swine, environment, equine
IRD
GISAID
Figure 1: Inconsistent host labels between IRD and GISAID databases: the intersection of hosts in the IRD and GISAID
databases is indicated in light green.
Figure 2: Data distribution (hosts).
3.3.2 Transformer
Transformer is an impactful neural network archi-
tecture developed in 2017 (Vaswani et al., 2017).
It was originally designed for machine translation,
but can be extended to other domains, such as solv-
ing protein folding problems (Grechishnikova, 2021).
Transformer lays the foundation for the development
of some state-of-the-art natural language processing
models, such as BERT (Devlin et al., 2018), T5 (Raf-
fel et al., 2019), and GPT-3 (Brown et al., 2020). One
of the biggest advantages of Transformer over tradi-
tional RNNs is that Transformer can process data in
parallel. Therefore, the Transformer can use GPUs to
speed them up and handle large text sequences well.
The innovations of Transformer neural network
include positional encoding and self-attention mech-
anism. Positional encoding stores the word order in
the data and helps the neural network to learn the or-
der information. The attention mechanism allows the
model to decide how to translate a word from the orig-
inal text to the output text. The self-attention mech-
anism, as the name suggests, pays attention to itself.
The self-attention mechanism allows the neural net-
work to understand the underlying meaning of words
in context by looking at the words around them. With
self-attention, neural networks can not only distin-
guish words but also reduce the amount of compu-
tation.
3.3.3 Convolutional Neural Network
A convolutional neural network (CNN) is typically
used to process images and achieves great success.
The idea of CNN is inspired by the visual process-
ing mechanism of the human brain, that is, neurons
are only activated by different features of the image,
such as edges. Two kinds of layers are often used in
CNNs, convolution layers and pooling layers. Con-
volution layers are the heart of CNNs, they imple-
ment convolution operators on the input image and fil-
ters. Pooling layers downsample the image to reduce
the learnable parameters. In this study, we use one-
dimensional convolution layers to process sequence
data.
3.4 Implementation and Evaluation
Methods
All models are built with Keras, trained on pre-19 data
sets, and tested on post-19 and incomplete data sets.
We apply transfer learning when it comes to incom-
plete data set. The architecture of the multi-channel
neural network architecture is shown in Fig. 5. The
Transformer architecture used in this study is the en-
coder shown in (Vaswani et al., 2017), we use 3 heads
and an input embedding with 32 dimensions.
End-to-End Multi-channel Neural Networks for Predicting Influenza a Virus Hosts and Antigenic Types
43
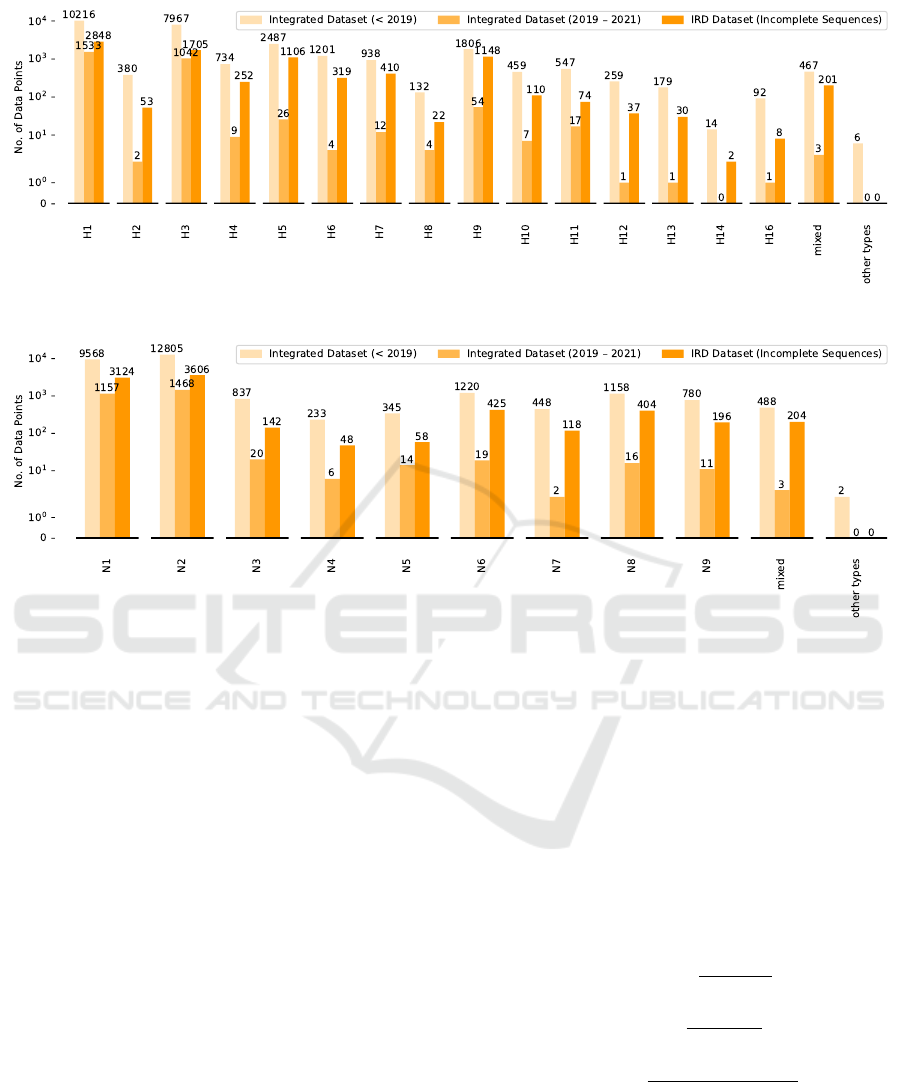
Figure 3: Data distribution (HA subtypes).
Figure 4: Data distribution (NA subtypes).
Some studies confuse the role of validation and
test sets, so they tune the model’s hyperparameters
on the testing set instead of a separate validation set.
This involves the risk of data leakage and undermines
the credibility of the results. Therefore, in contrast to
classic K-fold cross validation (CV), which split data
into training and testing set, nested CV uses an outer
CV to estimate the unbiased generalised error of the
model, and an inner CV for model selection or hyper-
parameter tuning. The outer CV splits the data into
training
outer
set and testing set, and the inner CV splits
the training
outer
set into training
inner
set and validation
set. The model is trained only on the training
inner
set,
tunes its hyperparameters based on its performance
on validation set, and tests its general performance on
testing set. In this study, the outer fold k
outer
is chosen
as 5 and inner fold k
inner
is 4. Fig. 6 shows the process
of building CV ensemble models.
The data sets used in this study are highly im-
balanced, and common evaluation measurements,
such as accuracy and receiver operating characteris-
tic (ROC) curve, can be misleading (Akosa, 2017),
(Davis and Goadrich, 2006). Precision-recall curve
(PRC), on the other hand, is more informative when
dealing with a highly skewed dataset (Saito and
Rehmsmeier, 2015), and has been widely used in re-
search (Bunescu et al., 2005), (Bockhorst and Craven,
2005), (Goadrich et al., 2004), (Davis et al., 2005).
It is unsuitable, however, if using linear interpolation
to calculate the area under the precision-recall curve
(AUPRC) (Davis and Goadrich, 2006). A better al-
ternative way, in this case, is average precision (AP)
score (Su et al., 2015). Besides, we also apply com-
mon evaluation metrics, i.e. precision, recall and F
1
score. The formulas of these evaluation metrics are
shown above:
Precision =
T P
T P + FP
(1)
Recall =
T P
T P + FN
(2)
F
1
= 2 ×
Precision × Recall
Precision + Recall
(3)
AP =
∑
n
(Recall
n
− Recall
n−1
Precision
n
) (4)
where TP, FP, TN, FN stand for true positive, false
positive, true negative and false negative. If positive
data is predicted as negative, then it counts as FN, and
so on for TN, TP and FP.
KDIR 2022 - 14th International Conference on Knowledge Discovery and Information Retrieval
44

NA seq
(1,200)
HA seq
(1,300)
Embedding
(1,200,100)
Embedding
(1,300,100)
Dense+Relu
(1,128)
Dense+Relu
(1,32)
Dense+Relu
(1,32)
Dense+Softmax
(1,44)
Dense+Softmax
(1,17)
Dense+Softmax
(1,11)
Concatenate
(1,500,100)
Hosts
HA SubtypesNA Subtypes
GRU
(1,500,64)
Dense+Relu
(1,512)
BiLSTM
(1,500,64)
Flatten
Dense+Relu
(1,512)
Dense+Relu
(1,256)
Conv
(1,500,256)
MaxPooling
(1,166,256)
Conv
(1,166,128)
MaxPooling
(1,55,128)
Conv
(1,55,64)
MaxPooling
(1,18,64)
Dropout
(1,18,64)
Flatten
BiLSTM
(1,500,32)
GRU
(1,500,32)
Flatten
Dense+Relu
(1,256)
Figure 5: The multi-channel neural network architecture.
Protein Sequences
Model 1Model 2Model 3Model 4
Out-of-Fold Predictions
Final Predictions
soft voting
split data into k
outer
fold
split data into k
inner
fold
Training
Testing
Validation
Figure 6: The process of building a CV ensemble.
4 RESULTS
The overall performance of the model tested on each
data set is shown in Fig. 7 to Fig. 9. Metrics like
AP are designed for binary classification but can be
extended to multi-class classification by applying a
one-vs-all strategy. This case entails taking one class
as positive and remaining as negative. We compare
each model with a baseline model, Basic Local Align-
ment Search Tool (BLAST), with default parame-
ters, in terms of AP, F
1
score, precision and recall
values. Five-fold cross-validation is also applied to
BLAST. The results of BLAST are framed by the
solid black line. All models outperform baseline,
especially multi-channel BiGRU and multi-channel
CNN, and the host classification task is harder than
the subtype classification task for all models.
All models are trained only on the pre-19 data
set and tested on the post-19 and incomplete data
sets. The pre-19 data set includes 44 hosts, 17
HA, and 11 NA, which is more diverse than post-19
set (15 hosts, 15 HA, and 10 NA) and the incom-
plete set (30 hosts, 16 HA, and 10 NA). Pre-19 and
post-19 data sets contain only complete sequences,
as opposed to the incomplete data set. Therefore,
the post-19 data set is less diverse, and all mod-
els performed better on the post-19 data set than
on the pre-19 and incomplete data sets, with the
best model being the multi-channel BiGRU. Multi-
channel BiGRU achieves 98.92% (98.88%, 98.97%)
AP, 98.33% (98.22%, 98.44%) precision, 98.13%
(98.05%, 98.22%) F
1
and 98.08% (97.98%, 98.18%)
recall on post-19 set.
When it comes to pre-19 and incomplete data sets,
multi-channel CNN yields best results, with an AP
of 93.38% (93.04%, 93.72%), a precision of 92.40%
(91.99%, 92.81%), a F
1
of 92.00% (91.57%, 92.44%)
and an recall of 93.01% (92.63%, 93.38%) on pre-
19 data set; and an AP of 96.41% (96.08%, 96.74%),
a precision of 93.65% (93.25%, 94.05%), a F
1
of
93.42% (93.04%, 93.81%) and an recall of 94.08%
(93.70%, 94.46%) on incomplete data set.
We further select two pairs of HA and NA
sequences from two strains that respectively indi-
cate that humans were infected with the first cases
of H5N8 and H10N3. A male patient was di-
agnosed with an A/H10N3 infection on 28 May
2021, and the isolated virus strain was named
as A/Jiangsu/428/2021. Whole-genome sequencing
analysis and phylogenetic analysis demonstrated that
this strain is of avian origin. More specifically, the
HA, NA, PB2, NS, PB1, MP, PA and NP genes of
this strain were closely related to some strains isolated
from chicken (Wang et al., 2021), which is aligns with
our model’s prediction, as shown in Table 2. The
second strain was isolated from poultry farm work-
End-to-End Multi-channel Neural Networks for Predicting Influenza a Virus Hosts and Antigenic Types
45

Figure 7: Comparison of Overall Performance Between Models (Hosts): the baseline results with BLAST are framed by the
black solid line.
Figure 8: Comparison of Overall Performance Between Models (HA subtypes): the baseline results with BLAST are framed
by the black solid line.
ers in Russia during a large-scale avian virus out-
break and was named A/Astrakhan/3212/2020. Phy-
logenetic analysis shows that this strain has high sim-
ilarity with some avian strains at the amino acid level
(Pyankova et al., 2021), which also matches our find-
ings.
5 CONCLUSION AND
DISCUSSION
Influenza viruses mutate rapidly, leading to seasonal
epidemics, but they rarely cause pandemics. How-
ever, influenza viruses can exacerbate underlying dis-
KDIR 2022 - 14th International Conference on Knowledge Discovery and Information Retrieval
46

Figure 9: Comparison of Overall Performance Between Models (NA Subtypes): the baseline results with BLAST are framed
by the black solid line.
Table 2: Case Study.
Algorithms Predicted Hosts Predicted HA Predicted NA
A/Jiangsu/428/2021
(human; H10N3)
BiGRU
chicken (0.7)
duck (0.3)
H10
N3 (0.95)
mixed (0.05)
BiLSTM
chicken (0.65)
duck (0.35)
H10 N3
CNN
chicken (0.6)
duck (0.4)
H10 (0.95)
mixed (0.05)
N3 (0.95)
mixed (0.05)
Transformer
duck (0.7)
mallard (0.25)
chicken (0.05)
H10
N3 (0.95)
mixed (0.05)
A/Astrakhan/3212/2020
(human; H5N8)
BiGRU
chicken (0.5)
duck (0.35)
goose (0.1)
environment (0.05)
H5 N8
BiLSTM
chicken (0.75)
duck (0.2)
swan (0.05)
H5 N8
CNN
chicken (0.6)
duck (0.2)
goose (0.2)
H5 N8
Transformer
duck (0.65)
chicken (0.25)
goose (0.1)
H5 N8
eases which increase the mortality risk. In this pa-
per, we have proposed multi-channel neural networks
that can rapidly and accurately predict viral hosts at a
lower taxonomical level as well as predict subtypes of
IAV given the HA and NA sequences. In contrast to
handcrafting the encoding scheme for transferring the
protein sequences to numerical vectors, our network
can learn the embedding of protein trigrams (three
consecutive amino acids in the sequence). This can
transfer a protein sequence to a dense vector. The
neural network architecture is designed to be multi-
channel, which takes multiple inputs and generates
multiple outputs, eliminating the need to train sepa-
rate models for similar tasks.
End-to-End Multi-channel Neural Networks for Predicting Influenza a Virus Hosts and Antigenic Types
47

We incorporate CNN, BiLSTM, BiGRU, and
Transformer algorithms as part of our multi-channel
neural network architecture, and we find that BiGRU
produces better results than other algorithms. A sim-
ple case study conducted in this study showed that our
results matched amino acid-level phylogenetic anal-
ysis in predicting the host and subtype of origin for
the first human cases of infection with H5N8 and
H10N3. Our study enables accurate prediction of po-
tential host origins and subtypes for this strain and
could benefit many resource-poor regions where ex-
pensive laboratory experiments are economically dif-
ficult to be conducted. However, as we only utilized
protein sequence data, it cannot predict the type of re-
ceptor that the virus may be compatible with. There-
fore, further research is needed to predict potential
viruses that are cross-species transmissible.
Furthermore, we only apply supervised learning
algorithms in this study, which rely on correctly la-
belled data and favour the majority of data, resulting
in the poor predictive ability for labels with insuffi-
cient data. Therefore, leveraging insufficient data is
also a goal of future research.
ACKNOWLEDGMENTS
The work is supported by University of Liverpool.
REFERENCES
Ahsan, R. and Ebrahimi, M. (2018). The first implication of
image processing techniques on influenza a virus sub-
typing based on ha/na protein sequences, using convo-
lutional deep neural network. bioRxiv, page 448159.
2
Akosa, J. (2017). Predictive accuracy: A misleading perfor-
mance measure for highly imbalanced data. In Pro-
ceedings of the SAS Global Forum, volume 12. 3.4
Asgari, E. and Mofrad, M. R. (2015). Continuous
distributed representation of biological sequences
for deep proteomics and genomics. PloS one,
10(11):e0141287. 3.2
Asha, K. and Kumar, B. (2019). Emerging influenza d virus
threat: what we know so far! Journal of Clinical
Medicine, 8(2):192. 1
Attaluri, P. K., Chen, Z., and Lu, G. (2010). Applying
neural networks to classify influenza virus antigenic
types and hosts. In 2010 IEEE Symposium on Com-
putational Intelligence in Bioinformatics and Compu-
tational Biology, pages 1–6. IEEE. 2
Attaluri, P. K., Chen, Z., Weerakoon, A. M., and Lu, G.
(2009). Integrating decision tree and hidden markov
model (hmm) for subtype prediction of human in-
fluenza a virus. In International Conference on Multi-
ple Criteria Decision Making, pages 52–58. Springer.
2
Bockhorst, J. and Craven, M. (2005). Markov networks
for detecting overlapping elements in sequence data.
Advances in Neural Information Processing Systems,
17:193–200. 3.4
Brown, T., Mann, B., Ryder, N., Subbiah, M., Kaplan, J. D.,
Dhariwal, P., Neelakantan, A., Shyam, P., Sastry, G.,
Askell, A., et al. (2020). Language models are few-
shot learners. Advances in neural information pro-
cessing systems, 33:1877–1901. 3.3.2
Bunescu, R., Ge, R., Kate, R. J., Marcotte, E. M., Mooney,
R. J., Ramani, A. K., and Wong, Y. W. (2005). Com-
parative experiments on learning information extrac-
tors for proteins and their interactions. Artificial intel-
ligence in medicine, 33(2):139–155. 3.4
Cao, Y., Liu, Z., Li, C., Li, J., and Chua, T.-S. (2019).
Multi-channel graph neural network for entity align-
ment. arXiv preprint arXiv:1908.09898. 2
Casas-Aparicio, G. A., Le
´
on-Rodr
´
ıguez, I., Hern
´
andez-
Zenteno, R. d. J., Castillejos-L
´
opez, M., Alvarado-
de la Barrera, C., Ormsby, C. E., and Reyes-Ter
´
an, G.
(2018). Aggressive fluid accumulation is associated
with acute kidney injury and mortality in a cohort of
patients with severe pneumonia caused by influenza a
h1n1 virus. PLoS One, 13(2):e0192592. 1
Chen, Y., Wang, K., Yang, W., Qing, Y., Huang, R., and
Chen, P. (2020). A multi-channel deep neural network
for relation extraction. IEEE Access, 8:13195–13203.
2
Cho, K., van Merrienboer, B., Bahdanau, D., and Bengio,
Y. (2014). On the properties of neural machine trans-
lation: Encoder-decoder approaches. 3.3.1
Chrysostomou, C., Alexandrou, F., Nicolaou, M. A., and
Seker, H. (2021). Classification of influenza hemag-
glutinin protein sequences using convolutional neural
networks. arXiv preprint arXiv:2108.04240. 2
Clayville, L. R. (2011). Influenza update: a review of cur-
rently available vaccines. Pharmacy and Therapeu-
tics, 36(10):659. 1, 2
Davis, J., Burnside, E. S., de Castro Dutra, I., Page, D.,
Ramakrishnan, R., Costa, V. S., and Shavlik, J. W.
(2005). View learning for statistical relational learn-
ing: With an application to mammography. In IJCAI,
pages 677–683. Citeseer. 3.4
Davis, J. and Goadrich, M. (2006). The relationship be-
tween precision-recall and roc curves. In Proceed-
ings of the 23rd international conference on Machine
learning, pages 233–240. 3.4
Devlin, J., Chang, M.-W., Lee, K., and Toutanova, K.
(2018). Bert: Pre-training of deep bidirectional trans-
formers for language understanding. arXiv preprint
arXiv:1810.04805. 3.3.2
Eng, C. L., Tong, J. C., and Tan, T. W. (2014). Predicting
host tropism of influenza a virus proteins using ran-
dom forest. BMC medical genomics, 7(3):1–11. 2
England, P. H. (2020). Influenza: the green book, chapter
19. 1
Fabija
´
nska, A. and Grabowski, S. (2019). Viral genome
deep classifier. IEEE Access, 7:81297–81307. 2
KDIR 2022 - 14th International Conference on Knowledge Discovery and Information Retrieval
48

George, A., Mostaani, Z., Geissenbuhler, D., Nikisins, O.,
Anjos, A., and Marcel, S. (2019). Biometric face pre-
sentation attack detection with multi-channel convo-
lutional neural network. IEEE Transactions on Infor-
mation Forensics and Security, 15:42–55. 2
Goadrich, M., Oliphant, L., and Shavlik, J. (2004).
Learning ensembles of first-order clauses for recall-
precision curves: A case study in biomedical informa-
tion extraction. In International Conference on Induc-
tive Logic Programming, pages 98–115. Springer. 3.4
Graves, A., Beringer, N., and Schmidhuber, J. (2005).
Rapid retraining on speech data with lstm recurrent
networks. Technical Report IDSIA-09–05, IDSIA.
3.3.1
Graves, A. and Schmidhuber, J. (2005). Framewise
phoneme classification with bidirectional lstm and
other neural network architectures. Neural networks,
18(5-6):602–610. 3.3.1
Grechishnikova, D. (2021). Transformer neural network for
protein-specific de novo drug generation as a machine
translation problem. Scientific reports, 11(1):1–13.
3.3.2
Hochreiter, S. and Schmidhuber, J. (1997a). Long short-
term memory. Neural computation, 9(8):1735–1780.
3.3.1
Hochreiter, S. and Schmidhuber, J. (1997b). Lstm can solve
hard long time lag problems. Advances in neural in-
formation processing systems, pages 473–479. 3.3.1
Iuliano, A. D., Roguski, K. M., Chang, H. H., Muscatello,
D. J., Palekar, R., Tempia, S., Cohen, C., Gran, J. M.,
Schanzer, D., Cowling, B. J., et al. (2018). Esti-
mates of global seasonal influenza-associated respi-
ratory mortality: a modelling study. The Lancet,
391(10127):1285–1300. 1
James, S. H. and Whitley, R. J. (2017). Influenza viruses.
In Infectious diseases, pages 1465–1471. Elsevier. 1
Kerzel, M., Ali, M., Ng, H. G., and Wermter, S. (2017).
Haptic material classification with a multi-channel
neural network. In 2017 International Joint Confer-
ence on Neural Networks (IJCNN), pages 439–446.
IEEE. 2
Kincaid, C. (2018). N-gram methods for influenza host
classification. In Proceedings of the International
Conference on Bioinformatics & Computational Biol-
ogy (BIOCOMP), pages 105–107. The Steering Com-
mittee of The World Congress in Computer Science,
Computer . . . . 2
Kwon, E., Cho, M., Kim, H., and Son, H. S. (2020).
A study on host tropism determinants of influenza
virus using machine learning. Current Bioinformat-
ics, 15(2):121–134. 2
Lau, L. L., Cowling, B. J., Fang, V. J., Chan, K.-H., Lau,
E. H., Lipsitch, M., Cheng, C. K., Houck, P. M.,
Uyeki, T. M., Peiris, J. M., et al. (2010). Viral
shedding and clinical illness in naturally acquired in-
fluenza virus infections. The Journal of infectious dis-
eases, 201(10):1509–1516. 1
Mills, C. E., Robins, J. M., and Lipsitch, M. (2004).
Transmissibility of 1918 pandemic influenza. Nature,
432(7019):904–906. 1
Mock, F., Viehweger, A., Barth, E., and Marz, M. (2021).
Vidhop, viral host prediction with deep learning.
Bioinformatics, 37(3):318–325. 2
Pyankova, O. G., Susloparov, I. M., Moiseeva, A. A.,
Kolosova, N. P., Onkhonova, G. S., Danilenko, A. V.,
Vakalova, E. V., Shendo, G. L., Nekeshina, N. N.,
Noskova, L. N., et al. (2021). Isolation of clade 2.3.
4.4 b a (h5n8), a highly pathogenic avian influenza
virus, from a worker during an outbreak on a poul-
try farm, russia, december 2020. Eurosurveillance,
26(24):2100439. 4
Raffel, C., Shazeer, N., Roberts, A., Lee, K., Narang,
S., Matena, M., Zhou, Y., Li, W., and Liu, P. J.
(2019). Exploring the limits of transfer learning
with a unified text-to-text transformer. arXiv preprint
arXiv:1910.10683. 3.3.2
Saito, T. and Rehmsmeier, M. (2015). The precision-recall
plot is more informative than the roc plot when eval-
uating binary classifiers on imbalanced datasets. PloS
one, 10(3):e0118432. 3.4
Scarafoni, D., Telfer, B. A., Ricke, D. O., Thornton, J. R.,
and Comolli, J. (2019). Predicting influenza a tropism
with end-to-end learning of deep networks. Health
security, 17(6):468–476. 2
Shaw, M. and Palese, P. (2013). Orthomyxoviridae, p 1151–
1185. fields virology. 1
Sherif, F. F., Zayed, N., and Fakhr, M. (2017). Classifica-
tion of host origin in influenza a virus by transferring
protein sequences into numerical feature vectors. Int
J Biol Biomed Eng, 11. 2
Shu, Y. and McCauley, J. (2017). Gisaid: Global initia-
tive on sharing all influenza data–from vision to real-
ity. Eurosurveillance, 22(13):30494. 3.1.1
Squires, R. B., Noronha, J., Hunt, V., Garc
´
ıa-Sastre, A.,
Macken, C., Baumgarth, N., Suarez, D., Pickett, B. E.,
Zhang, Y., Larsen, C. N., et al. (2012). Influenza re-
search database: an integrated bioinformatics resource
for influenza research and surveillance. Influenza and
other respiratory viruses, 6(6):404–416. 3.1.1
Su, W., Yuan, Y., and Zhu, M. (2015). A relationship be-
tween the average precision and the area under the roc
curve. In Proceedings of the 2015 International Con-
ference on The Theory of Information Retrieval, pages
349–352. 3.4
Thireou, T. and Reczko, M. (2007). Bidirectional
long short-term memory networks for predicting
the subcellular localization of eukaryotic proteins.
IEEE/ACM transactions on computational biology
and bioinformatics, 4(3):441–446. 3.3.1
Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones,
L., Gomez, A. N., Kaiser, Ł., and Polosukhin, I.
(2017). Attention is all you need. Advances in neural
information processing systems, 30. 3.3.2, 3.4
Vemula, S. V., Zhao, J., Liu, J., Wang, X., Biswas, S., and
Hewlett, I. (2016). Current approaches for diagno-
sis of influenza virus infections in humans. Viruses,
8(4):96. 2
Wang, Y., Niu, S., Zhang, B., Yang, C., and Zhou, Z. (2021).
Withdrawn: The whole genome analysis for the first
End-to-End Multi-channel Neural Networks for Predicting Influenza a Virus Hosts and Antigenic Types
49

human infection with h10n3 influenza virus in china.
4
Watanabe, T. (2013). Renal complications of seasonal and
pandemic influenza a virus infections. European jour-
nal of pediatrics, 172(1):15–22. 1
Wilde, J. A., McMillan, J. A., Serwint, J., Butta, J.,
O’Riordan, M. A., and Steinhoff, M. C. (1999). Ef-
fectiveness of influenza vaccine in health care profes-
sionals: a randomized trial. Jama, 281(10):908–913.
1
Xu, B., Tan, Z., Li, K., Jiang, T., and Peng, Y. (2017). Pre-
dicting the host of influenza viruses based on the word
vector. PeerJ, 5:e3579. 2
Xu, Y. and Wojtczak, D. (2022). Dive into machine
learning algorithms for influenza virus host predic-
tion with hemagglutinin sequences. arXiv preprint
arXiv:2207.13842. 3.2
Yang, Y., Wu, Q., Qiu, M., Wang, Y., and Chen, X. (2018).
Emotion recognition from multi-channel eeg through
parallel convolutional recurrent neural network. In
2018 international joint conference on neural net-
works (IJCNN), pages 1–7. IEEE. 2
Yin, R., Zhou, X., Rashid, S., and Kwoh, C. K. (2020).
Hopper: an adaptive model for probability estima-
tion of influenza reassortment through host prediction.
BMC medical genomics, 13(1):1–13. 2
KDIR 2022 - 14th International Conference on Knowledge Discovery and Information Retrieval
50
