
Developing CAR-T Therapy for Treating B Cell Malignancies
Clara Xi Wang
1,†
, Haoyang Guo
2,*,†
, Hanqin Yang
3
and Beibo Kang
4
1
College of Arts and Sciences, New York University, New York, NY, 10003, U.S.A.
2
Eberly College of Science, Pennsylvania State University, University Park, PA, 16802, U.S.A.
3
Jinan Foreign Language School, Jinan, Shandong, 250108, China
4
BASIS International School Guangzhou, Guangzhou, Guangdong, 510663, China
Keywords:
CAR-T, PD-1 Inhibitory Receptors, Cytokine Release Syndrome, CD40 Ligand, T Cell Type Ratio.
Abstract:
B cell lymphoma is a type hematopoietic malignancy with an average incidence rate of 4.3%. While B cell
lymphoma is not as deadly as other solid tumors, aggressive lymphomas such as the diffuse large B cell
lymphoma (DLBCL) can be fatal due to its fast-spreading characteristic and high metastatic ability. To more
effectively target B-cell lymphomas, scientists have recently created the chimeric antigen receptor (CAR) that
can actively detect the CD19 antigens secreted by cancer cells and directly activate T cells without binding to
the major histocompatibility complex (MHC). However, over time, many cancer cells have also developed
several mechanisms to escape the detection of T cells and to inhibit their function, which can significantly
hamper the overall efficacy of traditional CAR-T therapy. Furthermore, traditional CAR-T therapies may also
cause severe side effects, such as the cytokine release syndrome (CRS) caused by an overproduction of
proinflammatory cytokines. In this study, we examined six current research articles that address these immune
escape mechanisms as well as the side effects caused by traditional CAR-T therapies. We propose an
experimental CAR-T therapy that combines the major findings from this primary research, which, if proven
feasible, can substantially improve the overall efficacy of CAR cancer immunotherapy while significantly
reducing damage caused by side effects.
1 INTRODUCTION
B cell lymphoma is a hematopoietic malignancy
characterized by the proliferation of abnormal B
lymphocytes (Swiner, 2020). Most cases of B cell
lymphoma belong to the category of non-Hodgkin
lymphomas (NHL) such as the fast growth DLBCL
and the indolent chronic lymphocytic leukemia
(CLL). CLL generally grows much slower than
aggressive types, but they are also less curable with
standard treatments and, if left untreated, can
potentially grow into a more aggressive form of
cancer. Additionally, even though 40-50% of all
patients with DLBCL can achieve complete
remission after therapy, 30-40% of patients relapse
within a short time and 10% develop refractory
DLBCL, a type of cancer that does not respond to any
type of treatment. Patients with relapsed/refractory
DLBCL are less responsive towards conventional
*
Corresponding author.
†
They are both first authors
cancer therapies, and even if receiving second
treatments with higher doses of chemotherapy and
stem cell transplants, r/r DLBCL patients have a 1-
year survival rate of only 28% (Raut, 2014). These
characteristics make B cell lymphoma more
dangerous compared to other cancers. As a result,
there exists a dire need for new cancer therapies that
specifically target B cell lymphoma.
Current cancer treatment options include surgery,
chemotherapy, and radiation therapy, as well as the
latest techniques such as the minimally invasive
interventional radiology and immunotherapy.
However, because chemotherapies may cause serious
side effects such as non-specific cytotoxicity and
generalized immune suppression, scientists have
recently been investigating different
immunotherapies that utilize the immune system
itself to offer long-term remission through
immunological memory. Among different cancer
immunotherapies, CAR-T therapy that involves the
Wang, C., Guo, H., Yang, H. and Kang, B.
Developing CAR-T Therapy for Treating B Cell Malignancies.
DOI: 10.5220/0012015100003633
In Proceedings of the 4th International Conference on Biotechnology and Biomedicine (ICBB 2022), pages 149-155
ISBN: 978-989-758-637-8
Copyright
c
2023 by SCITEPRESS – Science and Technology Publications, Lda. Under CC license (CC BY-NC-ND 4.0)
149
manipulation of T lymphocytes in the adaptive
immune system has shown significantly greater
efficacy against B cell lymphoma in multiple
research studies and clinical trials.
1.1 CAR-T Therapy
The activation process of T lymphocytes involves the
interaction between the T cell receptor (TCR) and
specific antigens presented by the MHC as well as
several co-stimulation factors such as CD28, CD137,
and OX40. However, to evade T-cell detection tumor
cells have developed several immune inhibitory
functions, such as upregulating inhibitory molecules
as well as reducing MHC expression (Yilmaz, 2020).
CAR-T therapy addresses these limitations by
creating T cells that function independently of MHC
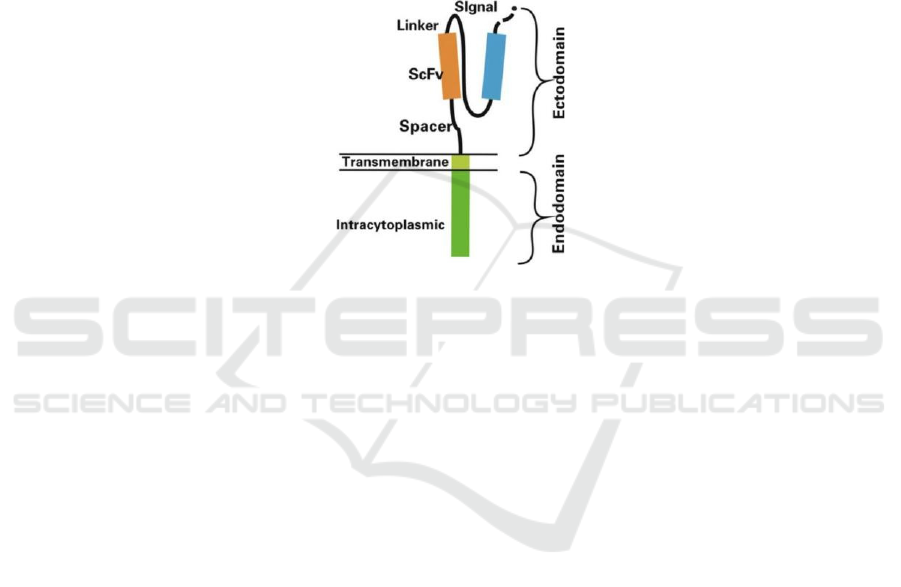
molecules (Graham, 2018). CARs are split into three
domains: ectodomains, a transmembrane domain,
and an intracellular domain of CD3ζ for signal
transduction (Figure 1). The ectodomain is the most
vital and different from traditional TCRs; it consists
of a signal peptide and the antigen recognition
domain of the single-chain Fragment variant (ScFv)
derived from the F
ab
region of antibodies fused by a
linker.
Figure 1: Structure of a Chimeric Antigen Receptor.
The Ectodomain consists of the linker, spacer,
ScFV head, and signal joint. The single-chain
variable fragment (ScFv) is responsible for the
specific antigen recognition from CAR cells.
Composed of the Fab regions of light and heavy
chains of an immunoglobulin, the ScFV is connected
to the CAR via a short linker peptide. The spacer, also
known as the hinge region, connects the antigen
recognition region to the surface of the membrane. It
enhances ScFV flexibility and promotes the binding
of recognition regions to target cells (Guedan, 2019).
Advantages of CAR-T arises from the fact that
they are MHC-independent, which allows it to
recognize any type of surface antigen, including
carbohydrates and lipids. Additionally, as memory T
cells remain in circulation long-term, the benefits of
CAR-T therapy can last for several years. CAR-T
therapies have little time for treatment, as it consists
of a single injection after which the patient can be
released after two weeks. Because the treatment itself
is not as aggressive as chemotherapy and radiation,
patients tend to have a much more rapid recovery.
Additionally, CAR-T can be a final resort for patients
who do not qualify for stem cell transplantation or
suffer from multiple relapses.
1.2 Side Effects of CAR-T Therapy
To date, CAR-T cells have shown remarkable
antitumor activity in patients because of the long-
lasting remissions in hematologic malignancies that
are not responding to standard therapies. For
example, the CAR-T therapy Axicabtagene
Ciloleucel can reach a tumor objective response rate
of 82% and a complete response rate of 54% (Levine,
2016). However, CAR-T therapies also have serious
limitations, such as the CRS. CRS is a severe side
effect of CAR-T therapies, in which the signalling
mechanism involved can provoke secretion of
cytokine and activation of macrophages (Neelapu,
2017). As a result, while investigating mechanisms to
decrease this side effect in CAR-T therapies,
researchers have also started to incorporate natural
killer (NK) cells and nanobodies in CAR-Therapies.
2 PRIMARY RESEARCH
STUDIES
To resolve different immune escape mechanisms
developed by tumor cells, we first examined two
papers, which introduced the idea of engineering
ICBB 2022 - International Conference on Biotechnology and Biomedicine
150

CAR-T cells to make CAR receptors actively secrete
anti-PD-1 antibody and CD40 ligand (CD40L).
Consequently, we evaluate the efficacy of the IL-6
binding protein in reducing the CRS discussed in the
third study. To maximize the overall effectiveness of
CAR-T therapy, we further examined the effector
function of different subsets of T cells discussed in
the fourth paper.
2.1 Engineered CAR-T Cells with
Self-Secreting Anti-PD1 Antibodies
Have Shown Optimistic Results in
Suppressing PD-1 Inhibitory
Receptors in the Tumor
Microenvironment (TME) of
CD-19 Expressing Tumors
CAR-T treatments’ long-term efficacy has been
impeded by the upregulation of several cell surface
inhibitory molecules in the TME, such as cytotoxic T
lymphocyte-associated protein (CTLA-4) and
programmed death-1 (PD-1). These inhibitory
receptors can negatively regulate the proper
activation of T cells by competing with the co-
stimulator CD28 for the B7 ligand. Without proper
activation, the overall immune response generated by
the adaptive immune system will be largely
hampered (Li, 2017). Among different inhibitory
molecules, the upregulation of PD-1 in CAR-T cells
can cause not only hypofunction of CAR-T cells but
also dysfunction of tumor infiltrate cells (TLC)
following specific antigen stimulation. As a result, in
this study, scientists aimed to combine the CAR-T
therapy with the PD-1 blockade mechanism to
overcome the inhibitory effect of PD-1. They
conducted the experiment using lung cancer line
NCI-H292 and designed a special CAR-T cell,
CAR19.alphaPD1, targeting CD-19 antigen by
inserting a gene fragment that can express the ScFv
from anti-PD1 antibody using retroviral vector. To
test whether CAR19.alphaPD1 can reduce the
immune inhibitory effect caused by PD-1,
researchers performed a competitive binding and
blocking assay on both CAR-T and
CAR19.alphaPD1 cells. In this experiment, T cells’
activity after being stimulated by anti-CD-3
antibodies was measured by intracellular IFN gamma
in the assay. According to the results, after the
recombinant human PD-L1 ligand were applied to
each well in the assay, the IFN gamma level
significantly, but this reduction of IFN gamma count
was quickly reversed after the addition of
CAR19.alphaPD1, indicating a successful blockade
of PD1 receptor by the self-secreted anti-PD-1
antibody. In addition, although PD-1 was upregulated
in both CAR-T and CAR19.alphaPD1 following
antigen stimulation, researchers in this study have
also discovered that expression of PD-1 was
significantly lower in CAR19.alphaPD1 T cells
compared to parental T cells, which can further
reduce the inhibitory effects caused by PD-1 in the
TME. Furthermore, in multiple other tests on
CAR19.alphaPD1’s ability to enhance antigen-
specific immune response, antitumor reactivity, T
cell proliferation, and T cell effector function,
CAR19.alphaPD1 in these tests all showed improved
efficacy compared to the control.
Based on the results, CAR19.alphaPD1 has
proven to be more effective in targeting H292-CD19
bearing tumors as well as reducing the immune
inhibitory signal in the TME compared to parental
CAR-T cells. Overall, CAR19.alphaPD1 can be
considered as an effective way to target CD19-
expressing tumors in future cancer immunotherapies.
2.2 Engineered CAR-T Cells with the
Ability to Secrete CD40L Can
Effectively Prevent Tumor Immune
Escape Caused by Antigen Loss
Tumor immune escape can occur via antigen loss so
that CAR-T cells lose their targets. To overcome
these negative impacts, CAR-T cells are engineered
to constitutively express CD40L to increase the
activation of the CD40/CD40L pathways in B cell
lymphoma. CD40 receptors are located on the surface
of abnormal B cells in B lymphoma. By transiently
activating CD40, CD40L can direct anti-proliferation
and apoptosis signals to cancer cells, thus effectively
reducing the outgrowth of B lymphoma (Odorizzi,
2012).
In addition, the CD40/CD40L pathway is highly
utilized for the activation of APCs, such as dendritic
cells (DC). The recruited APCs can further initiate
antitumor T cell responses by activating CD4
+
and
CD8
+
as well as secreting IL-12, which functions by
inhibiting the suppressive function of immune-
inhibitory macrophages, enhancing the antitumor
response of CAR-T cells, and recruit other non-CAR-
T cells at the same time (Elgueta, 2009).
Because the CD19
-
tumor can escape lysis by
traditional anti-CD19 CAR-T cells used in this study
(m1928z), the effectiveness of engineered CAR-T
cells with CD40L (m1928z-CD40L) on CD19
-
tumor
cells needs to be investigated. In this research study,
scientists modified the CD19
+
A20 lymphoma cell
line with the green fluorescent protein (GFP) and co-
Developing CAR-T Therapy for Treating B Cell Malignancies
151

cultured CD19
+
GFP
+
A20 cells with both m1928z
CAR-T cells and m1928z-CD40. The results showed
that by day 21, the outgrowth of CD19
-
tumor cells
can be successfully eliminated by m1928z-CD40L
compared to m1928z, indicating that m1928z-
CD40L is more effective in detecting antigen-
negative tumor cells in the long term through the
increased activation of the CD40/CD40L pathway.
(Elgueta, 2009).
2.3 CAR–T Cells with Mbail6 Have
Complete Antitumor Activity and
Neutralize Macrophage-Derived
IL-6, Which Could Prevent CRS
The CRS has been one of the most severe side effects
of traditional CAR-T therapy since the development
of the first generation of CAR-T cells.
Pathophysiologically, CRS is mediated by
proinflammatory cytokine interleukin-6 (IL-6)
mainly secreted by activated macrophages (Kuhn,
2019). Patients with CRS experience acute systemic
inflammatory responses characterized by fever,
fatigue, and headaches, which can be life-threatening
in some cases. Currently, the IL-6 receptor inhibitor
tocilizumab has been approved by FDA to treat CRS,
yet its effect is not stable. In this study, researchers
use a non-signaling membrane-bound IL-6 receptor
(mbaIL6) that possesses anti-CRS activity while
maintaining CAR-T cells’ complete antitumor
capacity. Compared with the control groups, which
are Jurkat cells with only GFP expression, mbaIL6-
expressing Jurkat cells showed a significant
reduction of IL-6 concentration in cell culture. The
neutralization of IL-6 by mbaIL6 is further tested
using the U937 cell line, in which stimulation is
dependent on IL-6-mediated STAT3
phosphorylation. When cocultured with mbaIL6-
expressing Jurkat cells, STAT phosphorylation
reaction was significantly reduced compared to the
control group. These results imply a successful
neutralization of IL-6 by mbaIL6, which is critical in
reducing CRS in CAR-T therapies.
Consequently, researchers engineered traditional
CAR-T cells using a bicistronic MSCV vector
containing genes encoding mbaIL6 and anti–CD19-
41BB-CD3z CAR. The mabIL6 expression in
peripheral blood T lymphocytes was proved to have
no interference with the immunophenotype of T cells.
Further experiments have also shown that the
expression of mabIL6 on anti-CD-19 CAR-T cells
does not affect their normal proliferation rate,
suggesting that a combination of anti-CD-19 CAR
and mabIL6 could be a frontline treatment for B-
lymphoid malignancies and multiple myeloma
(Neelapu, 2017).
2.4 Using a Defined Subset of CD8+
TCM and CD4+ TN Cells in a 1:1
Ratio Has Synergistic Antitumor
Effects in CAR-T Therapy
Traditional CAR-T therapy involves the injection of
CD3
+
CAR-T cells with nonspecific ratios of CD8
+
and CD4
+
subsets, leading to varying frequencies of
CD8
+
and CD4
+
T cells in all patients. Due to this
discrepancy, it is difficult to set a consistent baseline
for gauging the effectiveness of CAR-T therapy as
well as maximizing the positive effects of CAR-T. In
this study, subsets of T cells were tested for their
respective antitumor efficacy to determine the best
combination of specific T cells to maximize CAR-T
efficacy.
In this study, CD8
+
and CD4
+
were first tested for
subset efficacy and then for the combined effect of
superior CD8
+
and CD4
+
subsets on murine B cell
lymphomas in a set ratio (Sommermeyer et al., 2016).
CD8
+
and CD4
+
subsets were tested respectively in
NOD SCID IL2RgNULL (NSG) mice. The NSG
mice engrafted with CD19
+
Raji tumors then tested
for cytokine release, cytolytic abilities, and survival
grafts, with mice grafted with EGFRt-T cells serving
as control. The results revealed a hierarchy of subset
effector functions. CD8
+
T
CM
and T
N
cells showed
more cytolytic ability while CD4
+
T
N
cells had
superior cytokine release. Given that CD8
+
T
CM
and
CD4
+
T
N
had superior antitumor responses, doses of
either CD8
+
T
CM
, CD4
+
T
N
, or both of them combined
in a 1:1 ratio were administered to the mice. Mice that
received a 1:1 of CD8
+
T
CM
and CD4
+
T
N
showed
better survival rates as well as more complete tumor
eradication, judging from the bioluminescence
imaging data and survival curve. This data leads to
the conclusion that certain defined T cell subsets do
enhance antitumor response, in this case, a 1:1
combination of CD8
+
T
CM
and CD4
+
T
N
.
This study is significant because the antitumoral
efficacy of CAR-T cell subsets has never been
previously studied, and it is revealed that subsets of
CD8
+
T
CM
and CD4+ CAR-TN have the strongest
antitumoral effects. Moreover, this study
demonstrates combining CD8+ TCM and CD4+ TN
cells in a 1:1 ratio has a synergistic antitumor effect
than exclusively using one subset (Sallusto, 1999).
ICBB 2022 - International Conference on Biotechnology and Biomedicine
152

3 DISCUSSION
Upon further examination of current research studies,
we hypothesized a new experimental CAR-T therapy
to maximize CAR-T efficacy while reducing side
effects. This new CAR-T therapy will incorporate all
the modification processes shown in previous
research studies, including the self-secreting anti-PD-
1 CAR, the ability to secrete CD40 ligands and the
IL-6 binding proteins all administered in a 1:1 CD8
+
T
CM
to CD4
+
T
N
cells (figure 2), which, if proven to
be successful, can significantly increase the efficacy
of CAR immunotherapy while reducing the potential
side effects to the lowest level.
Figure 2: Cell model in the modified CAR-T cancer therapy.
CAR-T cells that target CD19 antigen (CAR19)
will be engineered to express anti-PD-1 antibody
(Anti-PD-1), CD40 ligand (CD40L), and membrane
binding IL-6 receptors (mbaIL6). These new proteins
are engineered on both CD8-expressing central
memory T cells (CD8
+
T
CM
) and CD4-expressing
naive T cells (CD4
+
T
N
), The ratio of the two types of
T cells will be manipulated to reach 1: 1 in this new
therapy.
To successfully create this new CAR-T therapy
targeting B cell malignancy as well as to test the
proper expression of different receptors and their
effectiveness, we propose several experiments using
different methods and analyzed potential results that
could reflect whether this new treatment is feasible
for future cancer therapies.
3.1 Gene Insertion During CAR-T
Engineering
The insertion of genes of anti-PD-1, CD40L, and
mbaIL6 achieved by introducing retroviral vectors
into T cells. The retroviral vector can accommodate
genes of interest and incorporate its genes into the
target cell genes (Hambach, 2020). It has been largely
used during CAR-T cell engineering because of its
high transfer efficiency as well as its variety of gene
expression based on different types of viruses used
(Guedan,, 2019). In our proposed experiments on
CAR engineering, the retroviral vector of MP71 will
be used to insert the ScFv of the anti-PD-1 antibody
derived from human mAb 5C4 (Habib, 2019); MSCV
vector will be used as the basis to transduce
membrane bond anti-IL-6 derived from human anti-
IL-6 monoclonal antibody AME-19a (Neelapu,
2017); finally, the SFG-m1928z-CD40L will be
constructed using Gibson Assembly (Elgueta, 2009)..
3.2 Testing Successful Expression of
Engineered Receptors Using
Western Blot
The successful expression of multiple receptors in
our proposed CAR-Ts can be detected using western
blot, a technique used to identify specific proteins of
interest through the binding of specially designed
antibodies. After binding to these designed
antibodies, proteins of interest will be stained and
visualized through gel electrophoresis. Different
types of proteins in a protein mixture will be
separated based on their molecular size (Kurian,
2020). Based on previous studies, it has been found
that CD40L has a molecular weight between 32 to 39
kDa (Odorizzi, 2012), while anti-PD-1-producing
CAR has a molecular weight of approximately 27kDa
(Habib, 2019). Since these two proteins have a
significant weight difference, antibodies designed
separately for these two proteins can be added
together in western blot using the original CD19
CAR as the control group. If the gel electrophoresis
result shows two distinct stains, one located on 27
Developing CAR-T Therapy for Treating B Cell Malignancies
153

kDa and the other between the range of 32 to 39 kDa
on the newly designed CAR-T cells while no clear
stain for the control group, it can be indicated that
CD40L and anti-PD-1-producing CAR has been
successfully expressed. If no stains are shown, or
they indicate different molecular weights, then the
engineered protein may not be successfully
expressed.
3.3 Detection and Isolation of Target
T cells Using Flow Cytometry
Assay
In this experiment, we aim to isolate CD8
+
central
memory T cells and CD4
+
naïve T cells based on their
surface marker proteins using flow cytometry assay.
As naïve T cells predominantly express CD62L
(Sallusto, 1999), we designed antibodies specifically
targeting CD4 and CD62L. After the flow cytometry
assay, cells showing higher affinity to both CD4 and
CD62L will be collected and used as the naïve T cell
in the therapy. Because central memory cells are have
CCR7+, the remaining memory T cells expressing
CCR7 will be further isolated using anti-CCR7,
completing isolation of CD4+ TN and CD8+ TCM.
After extracting T
CM
and T
N
cells, we propose to
further isolated T cells that simultaneously express all
three receptors engineered on the new CAR-T cell
using fluorescence-activated cell sorting (FACS).
The target T cells are identified and extracted based
on their binding intensity to antibodies designed for
the three different proteins. Based on previous
research, we have determined it is best to administer
the CD8
+
T
CM
cells and CD4
+
T
N
cells in a 1: 1 ratio,
which has shown to have the greatest synergistic
antitumor effect (Sallusto, 1999). Therefore, after
isolating the target T cell subsets, we will manipulate
a 1: 1 CD8
+
CAR-T
CM
to CD4
+
CAR-T
N
ratio before
conducting experiments in vivo.
3.4 Testing mbaIL-6’s Effective
Reduction of CRS
Infusion of CAR-T cells could lead to potentially life-
threatening CRS, which would sharply increase the
cost related to this treatment. Based on previous
experiments of mbaIL6, we decided to insert gene
encoding mbaIL6 to our newly designed ultimate
CAR-T cells to stay one step ahead of current studies
by combining mbaIL6 with other improvements on
CAR-T cells. Transduction of T lymphocytes could
be done with a construct that allows simultaneous
expression of mbaIL6, CAR, anti-PD1, and CD40L
to approach more effective antitumor capacity
without the presence of CRS. This construct could be
placed on an MSCV retroviral vector containing
genes of mbaIL6, anti-CD 19 CAR, anti-PD1, and
CD40L. For precise detection of anti-CD 19, CD19-
myc will be connected to the extracellular domain of
the CD19 molecule, and cells are stained with the
CD19-myc fusion protein and an anti-myc antibody.
Through variable control, degree of expression of
mbaIL6, CAR, anti-PD1, and CD40L, cell marker
profile (CD4, CD8, etc.), the proportion of naive,
effector, central memory, and effector memory, and
level of IL-6 Neutralization performed by mbaIL6
can be observed. To test whether IL-6 neutralization
by mbaIL6 can interfere with antitumor capacity
activated by CAR, we allow T cells to coculture with
CD19
+
OP-1 ALL cells and measure IFN-γ
production by flow cytometry assay after labeling
with anti-human IFN-γ–PE. The Level of cytotoxic
granules released by ultimate CAR-T cells can be
tested by staining with the anti-CD107a antibody.
The xenograft models could be used to determine the
in vivo antitumor capacity of ultimate CAR-T cells,
CD191 ALL cell line Nalm-6 IV (a B cell precursor
leukemia cell line) will be injected in NSG mice
along with ultimate CAR-T with and without
mbaIL6. Furthermore, levels of human IL-6
neutralization in vivo can be determined through
injecting ultimate CAR-T with and without mbaIL6
accompanied by IP injection of human IL-6 several
days later. If this combination works in xenograft
models, we would see considerable antitumor
capacity with and without expression of mbaIL6 on
ultimate CAR-T for a relatively long period.
Significantly, ultimate CAR-T expressing mbaIL6
can neutralize mbaIL6 to a lower level compared
with ultimate CAR-T not expressing mbaIL6 and
prevent CRS (Neelapu, 2017).
3.5 Testing T Cell Efficacy Through
Cytolytic Ability and Survival Rate
of Engrafted Mice
After injecting our proposed CAR-T therapy in the
NSG mice models, the efficacy of our modified
CAR-T therapy, we propose to use chromium release
assay as a measurement of cytolytic activity and
survival grafts to detect survival rates of mice bearing
CD-19 B lymphomas. Mice receiving only EGFRt-T
cells will serve as the control. If this combined
treatment were effective, we would expect to see a
significant increase in the survival rates and cytolytic
abilities of mice receiving the modified CAR-T
therapy compared to those receiving the control.
However, if the chromium release assay and survival
ICBB 2022 - International Conference on Biotechnology and Biomedicine
154

rates show no significant difference or show a
significant decrease between the modified CAR-T
group and the control, we can deduce that our
modified CAR-T therapy fails to have a synergistic
effect on tumor masses.
4 CONCLUSION
In this study, we looked into six current research on
various CAR cancer therapies. Based on the methods
and results shown in these research experiments,
proposed a new CAR-T therapy model that combines
anti-PD1 and CD40L secretion, mbaIL6 receptor. To
further improve the efficacy of this new model, we
also suggested an optimal 1:1 CD8 T
CM
to CD4 T
N
cell ratio. The proposed experiment will be tested in
vivo using the NSG mice model, which, if proven to
be successful, can significantly improve the overall
CAR-T cell efficacy in treating B cell lymphoma.
However, because we are unable to conduct actual
experiments, the feasibility and overall efficacy of
this newly designed CAR-T therapy will have to be
thoroughly investigated in future experiments.
REFERENCES
Elgueta, R., Benson, M. J., de Vries, V. C., Wasiuk, A.,
Guo, Y., & Noelle, R. J. (2009). Molecular mechanism
and function of CD40/CD40L engagement in the
immune system. Immunological Reviews, 229(1),
10.1111/j.1600-065X.2009.00782.x.
https://doi.org/10.1111/j.1600-065X.2009.00782.x
Graham, C., Hewitson, R., Pagliuca, A., & Benjamin, R.
(2018). Cancer immunotherapy with CAR-T cells –
behold the future. Clinical Medicine, 18(4), 324–328.
https://doi.org/10.7861/clinmedicine.18-4-324
Guedan, S., Calderon, H., Posey, A. D., & Maus, M. V.
(2019). Engineering and Design of Chimeric Antigen
Receptors. Molecular Therapy. Methods & Clinical
Development, 12, 145–156.
https://doi.org/10.1016/j.omtm.2018.12.009
Habib, S., Tariq, S. M., & Tariq, M. (2019). Chimeric
Antigen Receptor-Natural Killer Cells: The Future of
Cancer Immunotherapy. The Ochsner Journal, 19(3),
186–187. https://doi.org/10.31486/toj.19.0033
Hambach, J., Riecken, K., Cichutek, S., Schütze, K.,
Albrecht, B., Petry, K., Röckendorf, J. L., Baum, N.,
Kröger, N., Hansen, T., Schuch, G., Haag, F., Adam,
G., Fehse, B., Bannas, P., & Koch-Nolte, F. (2020).
Targeting CD38-Expressing Multiple Myeloma and
Burkitt Lymphoma Cells In Vitro with Nanobody-
Based Chimeric Antigen Receptors (Nb-CARs). Cells,
9(2), 321. https://doi.org/10.3390/cells9020321
Kuhn, N. F., Purdon, T. J., van Leeuwen, D. G., Lopez, A.
V., Curran, K. J., Daniyan, A. F., & Brentjens, R. J.
(2019). CD40 Ligand-Modified Chimeric Antigen
Receptor (CAR) T Cells Enhance Antitumor Function
by Eliciting an Endogenous Antitumor Response.
Cancer Cell, 35(3), 473-488.e6.
https://doi.org/10.1016/j.ccell.2019.02.006
Kurian, K. M., Watson, C. J., & Wyllie, A. H. (2000).
Retroviral vectors. Molecular Pathology, 53(4), 173–
176.
Levine, B. L., Miskin, J., Wonnacott, K., & Keir, C. (2016).
Global Manufacturing of CAR T Cell Therapy.
Molecular Therapy. Methods & Clinical Development,
4, 92–101.
https://doi.org/10.1016/j.omtm.2016.12.006
Li, S., Siriwon, N., Zhang, X., Yang, S., Jin, T., He, F.,
Kim, Y. J., Mac, J., Lu, Z., Wang, S., Han, X., & Wang,
P. (2017). Enhanced Cancer Immunotherapy by
Chimeric Antigen Receptor–Modified T Cells
Engineered to Secrete Checkpoint Inhibitors. Clinical
Cancer Research, 23(22), 6982–6992.
Neelapu, S. S., Locke, F. L., Bartlett, N. L., Lekakis, L. J.,
Miklos, D. B., Jacobson, C. A., Braunschweig, I.,
Oluwole, O. O., Siddiqi, T., Lin, Y., Timmerman, J. M.,
Stiff, P. J., Friedberg, J. W., Flinn, I. W., Goy, A., Hill,
B. T., Smith, M. R., Deol, A., Farooq, U., … Go, W. Y.
(2017). Axicabtagene Ciloleucel CAR T-Cell Therapy
in Refractory Large B-Cell Lymphoma. The New
England Journal of Medicine, 377(26), 2531–2544.
https://doi.org/10.1056/NEJMoa1707447
Odorizzi, P., & Wherry, E. J. (2012). Inhibitory Receptors
on Lymphocytes: Insights from Infections. Journal of
Immunology (Baltimore, Md. : 1950), 188(7), 2957–
2965. https://doi.org/10.4049/jimmunol.1100038
Raut, L. S., & Chakrabarti, P. P. (2014). Management of
relapsed-refractory diffuse large B cell lymphoma.
South Asian Journal of Cancer, 3(1), 66–70.
https://doi.org/10.4103/2278-330X.126531
Sallusto, F., Lenig, D., Förster, R., Lipp, M., &
Lanzavecchia, A. (1999). Two subsets of memory T
lymphocytes with distinct homing potentials and
effector functions. Nature, 401(6754), 708–712.
https://doi.org/10.1038/44385
Swiner, C. (2020, June 30). What Is B-Cell Lymphoma?
WebMD.
https://www.webmd.com/cancer/lymphoma/what-is-
b-cell-lymphoma.
Yilmaz, A., Cui, H., Caligiuri, M. A., & Yu, J. (2020).
Chimeric antigen receptor-engineered natural killer
cells for cancer immunotherapy. Journal of
Hematology & Oncology, 13(1), 168.
https://doi.org/10.1186/s13045-020-00998-9
Developing CAR-T Therapy for Treating B Cell Malignancies
155
