
Efficient Hashing of Multiple Spaced Seeds with Application
Eleonora Mian, Enrico Petrucci, Cinzia Pizzi and Matteo Comin
Department of Information Engineering, University of Padova, Padova, 35131, Italy
Keywords:
k-Mers, Gapped q-Gram, Multiple Spaced Seeds, Efficient Hashing.
Abstract:
Alignment-Free analysis of sequences has enabled high-throughput processing of sequencing data in many
bioinformatics pipelines. Hashing k-mers is a common function across many alignment-free applications and
it is widely used for indexing, querying and rapid similarity search. Recently, spaced seeds, a special type
of pattern that accounts for errors or mutations, are routinely used instead of k-mers. Spaced seeds allow to
improve the sensitivity, with respect to k-mers, in many applications, however the hashing of spaced seeds
increases substantially the computational time. Moreover, if multiple spaced seeds are used the accuracy can
further increases at the cost of running time. In this paper we address the problem of efficient multiple spaced
seed hashing. The proposed algorithms exploit the similarity of adjacent spaced seed hash values in an input
sequence in order to efficiently compute the next hashes. We report the results on several tests which show that
our methods significantly outperform the previously proposed algorithms, with a speedup that can reach 20x.
We also apply these efficient spaced seeds hashing algorithms to an application in the field of metagenomic,
the classification of reads performed by Clark-S (Ounit and Lonardi, 2016), and we shown that a significant
speedup can be obtained, thus resolving the slowdown introduced by the use of multiple spaced seeds. Code
available at: https://github.com/CominLab/MISSH.
1 INTRODUCTION
Alignment-free methods are at the basis of
many state-of-the-art tools in sequence analysis
(A.Zielezinski et al., 2017). In fact, the scale of
data produced by current sequencing technologies
is such that alignment-based approaches struggle to
cope with the high throughput processing needed by
current applications based on sequence analysis of
massive datasets.
Most alignment-free approaches are based on se-
quences decomposition into consecutive k-mers and
on their indexing through efficient data structures
(Marc¸ais et al., 2019). An example of a popular ap-
plication for such indexes is similarity search for se-
quence classification, where the query sequence is
also decomposed into k-mers and rapidly searched for
matches on the data structure to determine the closest
similarity with the indexed sequences. Kraken (Wood
and Salzberg, 2014) and CLARK (Ounit et al., 2015)
are two popular examples of k-mers based classifiers
for reads in metagenomics samples.
With respect to alignment-based techniques, k-
mer based approaches are orders of magnitude faster.
However, speed improvements come at the cost of
a loss of sensitivity, due to the exact matches re-
quired in all k positions of the patterns. To allevi-
ate this problem variants of the exact k -mer matches
paradigm have been proposed. Notable examples of
such generalizations are, for example, considering
longest matches with mismatches rather than fixed
length exact matches (Leimeister and Morgenstern,
2014; Apostolico et al., 2016) or allowing for not con-
secutive matches within k-mers.
The latter approach was first proposed in the con-
text of homology search with the tool PatternHunter
(Ma et al., 2002). In that paper, Ma and colleagues
introduced the concept of spaced seed, i.e. pat-
terns of fixed length that allow for wildcards in pre-
determined positions, greatly improving the chance of
finding relevant similarities. Besides further improve-
ments in homology search, e.g. (Kucherov et al.,
2006; No
´
e and Martin, 2014), spaced seeds have since
enabled the design of many successful algorithms in
several different context in bioinformatics. A non-
exhaustive list of examples includes: protein clas-
sification (Onodera and Shibuya, 2013); read map-
ping (Rumble et al., 2009); phylogenetic tree re-
construction (Leimeister et al., 2014; R
¨
ohling et al.,
2020); metagenomics reads clustering and classifica-
Mian, E., Petrucci, E., Pizzi, C. and Comin, M.
Efficient Hashing of Multiple Spaced Seeds with Application.
DOI: 10.5220/0011632900003414
In Proceedings of the 16th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2023) - Volume 3: BIOINFORMATICS, pages 155-162
ISBN: 978-989-758-631-6; ISSN: 2184-4305
Copyright
c
2023 by SCITEPRESS – Science and Technology Publications, Lda. Under CC license (CC BY-NC-ND 4.0)
155

tion (B
ˇ
rinda et al., 2015; Girotto et al., 2017b; Ounit
and Lonardi, 2016; Wood et al., 2019); and the predic-
tion of protein-protein interaction (Li and Ilie, 2017).
Although alignment-free techniques based on
spaced seeds are faster than alignment-based ap-
proaches, they suffer from a notable slowdown in run-
ning time with respect to equivalent k-mers based so-
lutions. Indeed, k -mers indexing can benefit from the
fact that consecutive k-mers share a large portion of
the sequence and this can be exploited for faster hash-
ing (Mohamadi et al., 2016). On the contrary, the
projection of two consecutive segments of a sequence
with respect to a given spaced seeds might have very
little in common, due to the positioning of the wild-
cards (Girotto et al., 2018b).
This phenomenon is amplified when multiple
spaced seeds are used. For example, it was reported
in Clark-S (Ounit and Lonardi, 2016) that using three
spaced seeds lead to a 17x slowdown. However, using
multiple spaced seed can further improve the results
of using a single spaced seed (Dencker et al., 2019).
These considerations motivate the compelling
need for the development of fast approaches for ef-
ficient hashing of (multiple) spaced seeds. The first
attempt to speeding up this process was done in (Har-
ris, 2007), where hard coding was used to speed-
up non-linear packing. More recently, several ap-
proaches have been developed based on block in-
dexing (Girotto et al., 2018a), and on spaced seeds
self correlation (Girotto et al., 2017a; Girotto et al.,
2018b; Petrucci et al., 2020) to reuse part of the hash
values that had already been computed for hashing the
previous segments of the sequence.
In this paper we present a series of algorithms
called ISSH Multi, a generalization of the ISSH
method (Petrucci et al., 2020), specifically designed
for multiple spaced seeds. Experimental comparison
showed that by processing multiple spaced seeds at
once, it is possible to further improve the speed of
spaced seed hashing. Moreover, we applied these ef-
ficient spaced seeds hashing algorithms to the metage-
nomic classification of reads performed by Clark-S
(Ounit and Lonardi, 2016), and we showed that a sig-
nificant speedup can be obtained in practice on a real
application.
2 METHODS: HASHING OF
MULTIPLE SPACED SEEDS
2.1 Spaced Seeds Hashing: Background
In this section we define what spaced seeds are and
describe the way they can be used to perform the
hashing of a DNA sequence, as well as highlighting
what makes the hashing of sequences based on k-mers
more computationally efficient.
A spaced seed Q is a string over the alphabet
{0, 1} of length k = s(Q) and weight w = |Q|. This
string contains w matching positions (corresponding
to the character ‘1’), and k − w non-matching posi-
tions, or “don’t care”, (corresponding to the character
‘0’): the spaced seed’s weight is therefore equal to the
number of 1s contained in the seed itself. A spaced
seed Q can be represented as a set of non negative in-
tegers corresponding to the matching positions (1s) in
the seed.
These spaced seeds can be used as a mask that,
when super-imposed on a DNA sequence with an
AND operation, only lets through the characters that
corresponds to a matching position ‘1’. The sub-
strings obtained will be called Q-grams, and they are
defined as x[i+Q] = {x
i+k
, k ∈ Q}, where i is the posi-
tion in the sequence where the spaced seed is aligned.
Example 2.1. Given the spaced seed Q = 10111011,
defined as Q = {0, 2, 3, 4, 6, 7}, with length k = 8
and weight w = 6. Let us consider the string x =
AT GGCAGTCA, the Q-gram x[1 + Q] = T GCATC
can be defined as follows:
x A T G G C A G T C A
Q 1 0 1 1 1 0 1 1
x[1+Q] T G C A T C
As mentioned before, the use of spaced seeds
significantly improves the sensitivity of similarity
searches regarding DNA sequences, but, when used
in conjunction with hashing, it does so at the detri-
ment of the computation speed. The reason for this
is that the hash of a generic k -mer can be computed
from the hash of its predecessor, something that is not
immediately possible when computing the hash of a
Q-gram obtained from a spaced seed.
In this paper, for ease of discussion, we will
consider as hashing function the simple encoding
of a string, that is a special case of the Rabin-
Karp rolling hash. Let’s consider a coding func-
tion from the DNA alphabet A = {A,C, G, T } to a
binary codeword, encode : A → {0, 1}
log
2
|A|
, where
encode(A) = 00, encode(C) = 01, encode(G) = 10,
and encode(T ) = 11. Given a string of n consecu-
tive characters, first the encoding function is applied
to each character. Then a shift based on the position
the character occupies in the original string will be
performed. Formally:
h(x[i + Q]) =
_
k∈Q
(encode(x
i+k
) ≪ m(k) ∗ log
2
|A|) (1)
where m(k) = |{i ∈ Q, such that i < k}| is the number
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
156

of matching positions that appear to the left of k. Each
character is encoded, as seen above, with 2 bits, and
the number of shifts required to set the k-th character
in the correct position is m(k) ∗log
2
|A|.
Example 2.2. Given the sequence x, we compute
the hashing value of the first Q-gram obtained using
spaced seed Q = 10111011. x
0
is the Q-gram obtained
from the first projection:
x = A T G G C A G T C A
1 0 1 1 1 0 1 1
x
0
= A G G C G T = x[0 + Q]
The hashing value of x
0
is therefore:
h(x
0
) = h(AGGCGT )
= 111001101000
To compute the hash of a contiguous k-mer it is
possible to use the hash of its predecessor. In fact,
given the hashing value at position i, the hashing for
position i + 1 can be obtained with two operations, a
shift and the insertion of the encoding of the new sym-
bol, since the two hashes share k − 1 symbols. How-
ever, if we consider the case of a spaced seed Q, we
can clearly see that this observation does not hold. In
fact, in the above example, two consecutive Q-grams,
like x[0 + Q] = AGGCGT and x[1 + Q] = T GCATC,
do not necessarily have much in common.
Computing efficiently the hash of the a DNA se-
quence based on a spaced seed is more complicated
than with k-mers. When we overlap the spaced seed
on the DNA sequence, we need, first of all, to ex-
tract the corresponding Q-gram, and only then we can
compute the hash of this substring. Then, the spaced
seed is moved one position to the right, and the pro-
cess is repeated for each Q-gram that can thus be ex-
tracted from the DNA sequence.
x
0
= A G G C G T
x
1
= T G C A T C
h(x
0
) = 11 10 0110 1000
h(x
1
) = 01 11 00 0110 11
It is evident that recovering certain positions from
the previous hash to reuse them in the second one
is not as straightforward as with k-mers, because the
spaced seed, depending on how the matching posi-
tions where overlapping on the DNA sequence, will
have filtered different nucleotides.
2.2 Previous Work
The problem of spaced seeds hashing is to find in-
creasingly more efficient ways to exploit the similar-
ity between different Q-grams, in order to minimize
the number of encoding and shift operations that need
to be applied to compute the hashing of a DNA se-
quence based on spaced seeds. Here, we review the
most common approaches proposed in the literature:
Fast spaced Seed Hashing (Girotto et al., 2018b),
Fast Indexing for spaced Seed Hashing (Girotto et al.,
2018a) and Iterative Spaced Seed Hashing (Petrucci
et al., 2020).
The first approach is Fast spaced Seed Hashing
(FSH) (Girotto et al., 2018b), which exploits the sim-
ilarity of adjacent hash values of the same DNA se-
quence to compute each hash more efficiently. To do
so, it recovers some information from previous com-
putations: specifically, it reuses parts of a hash value
already computed by extracting them through a mask
and then combining the result with the encoding of
the remaining positions.
In Fast Indexing for Spaced seed Hashing (FISH),
described in (Girotto et al., 2018a), a completely dif-
ferent approach, based on block-indexing, was pro-
posed. A unit block is a block of consecutive ‘1’s, in
which the spaced seed is decomposed. These blocks
are interpreted by the algorithm as k-mers of different
lengths, which can be hashed quickly. Since FISH re-
duces the problem of spaced seed hashing to the hash-
ing its k-mer components, this approach can obtain a
substantial improvement in computation time with re-
spect to FSH.
The Iterative Spaced Seed Hashing (ISSH) algo-
rithm described in (Petrucci et al., 2020), finally, is an
evolution of FSH. What distinguishes the two is that
ISSH recovers information from more than one pre-
vious hash: it uses a greedy approach to iteratively
search for hashes that allow to maximize the num-
ber of positions recovered (each time only taking into
consideration the characters remaining to be encoded)
and does not stop until either all positions are recov-
ered, or no more positions can possibly be recovered
from the hashes already computed.
Example 2.3. Given spaced seed Q = 11101010101,
we look for the positions where both Q and its shift
Q − 1 or Q − 2 present a matching position ‘1’. Those
positions in the hashing of the Q-gram extracted by
the seed Q will be recovered and reused to compute
the hashing of the Q-gram corresponding to the
spaced seed Q − 1 (or Q − 2).
Efficient Hashing of Multiple Spaced Seeds with Application
157

FSH only considers the first possible attachment
point:
Pos. ‘1’ 0 1 2 3 4 5 6
Q 1 1 1 0 1 0 1 0 1 0 1
Q − 1 1 1 1 0 1 0 1 0 1 0 1
Pos. ‘1’ 0 1 2 3 4 5 6
Q 1 1 1 0 1 0 1 0 1 0 1
Q − 2 1 1 1 0 1 0 1 0 1 0 1
ISSH can also choose a different shift, as seen
when recovering positions from Q − 2:
Pos. ‘1’ 0 1 2 3 4 5 6
Q 1 1 1 0 1 0 1 0 1 0 1
Q − 1 1 1 1 0 1 0 1 0 1 0 1
Pos. ‘1’ 0 1 2 3 4 5 6
Q 1 1 1 0 1 0 1 0 1 0 1
Q − 2 1 1 1 0 1 0 1 0 1 0 1
In FSH and ISSH masks are used to extract from
a previous hash the relevant positions to be recovered,
and these masks can be computed by a preprocessing
that only takes the spaced seeds in input, and is inde-
pendent from the actual DNA sequence to be hashed.
2.3 Efficient Multiple Spaced Seed
Hashing
In the following we describe our contribution to the
speedup of the computation of the hashing of DNA
sequences using multiple spaced seeds. We expand
on the method described in ISSH (Petrucci et al.,
2020) to further improve its efficiency by consider-
ing a group of spaced seeds at the same time, and we
analyze and compare three different approaches to do
so.
2.3.1 ISSH Multi
The first method we describe is called ISSH Multi.
This approach considers multiple spaced seeds at the
same time, but the hashing of the DNA sequence is
computed almost completely independently for each
spaced seed. This means that, for each hashing, infor-
mation is only recovered from hash values that were
calculated on the same spaced seed. In practice, this
implies that the preprocessing necessary for this ap-
proach is the same that was used in ISSH.
However, unlike ISSH, the hashing matrix is filled
in by columns: for each possible overlap with the
DNA sequence, the hash values are computed for each
ISSH Multi
Current hash
Encoding computed
only once
Previously computed hashes, more than one is used in order to
recover all the position but the last one.
In each row only previous hashes from the same row are reused
ISSH Multi Column
Current hash
Previously computed hashes, more than one is used in
order to recover all the positions
ISSH Multi Row
Previously computed hashes, more than one
is used in order to recover all the positions
Current hash
Figure 1: A schematic representation of the ISSH Multi
computations. The rows of the matrix represent the differ-
ent spaced seeds, whereas the columns the position of the
sequence where to compute the hash.
spaced seed one after the other. This provides a com-
putational advantage in the encoding of the last char-
acter of each Q-gram, which is always being seen for
the first time and which always belongs to all hash-
ing values, because by definition a spaced seed’s last
character is ‘1’. This encoding can therefore be com-
puted only once through the encoding function, and
inserted into the hash values of the current overlap for
all spaced seeds considered, thus saving a number of
encoding operations equal to # of spaced seeds−1 for
each position. For space limitation we do not report
the algorithm as it is a special case of the next algo-
rithm ISSH Multi Column. A schematic description
of the method ISSH Multi is shown in Figure 1.
2.3.2 ISSH Multi Column
The second approach we explored is ISSH Multi Col-
umn, which, once again, considers multiple spaced
seeds in input at the same time, and, like ISSH Multi,
it fills in the hashing matrix by columns.
The difference with the former is that ISSH Multi
Column introduces a new degree of freedom, by al-
lowing the choice of the hashing to recover position
from to include hashes computed using a different
spaced seed from the current one considered (that is, it
searches the best hash also from different rows/spaced
seeds of the hashing matrix). A schematic description
of ISSH Multi Column can be found in Figure 1.
This means that, to compute the generic hash
h(i, j), where i is the index of the spaced seed (i.e.
row in the hashing matrix) and j is the index of the
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
158

Algorithm 1: ISSH Multi Column (x, spacedSeeds,
ssLength, ssWeight).
Input: x ← DNA sequence
spacedSeeds ← group of spaced seeds
ssLength ← length of the spaced seeds
ssWeight ← weight of the spaced seeds
Output: Hash ← matrix containing all the computed
hashes.
1: for j := 0, |x| − ssLength do
2: for all i ∈ spacedSeeds do
3: Hash[i][ j] := 0
4: while missing positions can be recovered
from available hashes do
5: (n, m, l) := such that condition (2)
holds and Hash[n][m] with shift l allows to re-
cover the highest number of missing positions.
6: Hash[i][ j] := Hash[i][ j] OR
(Hash[n][m] ≫ 2 ∗ l AND mask(i, n, m, l))
7: end while
8: if there are still missing positions then
9: add missing encodings to Hash(i, j)
10: end if
11: end for
12: end for
13: return matrix Hash
Q-gram to be hashed (i.e. column in the hashing ma-
trix), we can search for the one that allows to recover
most positions among all previously computed hashes
h(n, m). Note, that the best previous hash h(n, m), it
does not depend on the sequence to be hashed, but
only on the structure of the spaced seed, and thus
the best values of (n, m) and the shift l can be eas-
ily pre-computed. In order to extract the symbols
from h(n, m) to be reused in the new hash h(i, j) we
define a mask, mask(i, n, m, l), that filters these po-
sitions. However, the hash h(n, m) must be already
computed. The following condition sums up all the
constraints that a hash h(n, m) needs to satisfy in or-
der to be used for recovering positions for the current
hash h(i, j):
(m < j OR (m = j AND n < i)) AND m ≥ 0 (2)
The big advantage of this method is that the num-
ber of encoding operations to be done during the tran-
sient is much lower: even the hashing of the first
Q-gram of the second spaced seed already has the
chance of recovering positions from the very first
hash, which wasn’t possible before. Ultimately, the
encoding function is only used once for each charac-
ter in the sequence even during the transient, allowing
for a significant improvement in computation times
compared to ISSH Multi.
2.3.3 ISSH Multi Row
The third and last method – ISSH Multi Row – fol-
lows the same scheme of the previous one, but it fills
in the hashing matrix by rows, making it possible
to also recover positions from hashes that have been
computed with different spaced seeds, and that corre-
spond to a Q-gram on the right of the current one.
Equivalently to what we described before, to com-
pute the generic hash h(i, j) we can search for a hash
h(n, m) from which to recover information where n <
i (that is, all preceding rows) or n = i and m < j (that
is, same row but preceding columns – all hashes that
have been computed with the same spaced seed but of
Q-grams extracted from preceding overlaps). There-
fore in order to use h(n, m) to compute h(i, j) the fol-
lowing condition must hold:
[n < i OR (n = i AND m < j)]
AND (0 ≤ m ≤ |sequence| − s(Q))
(3)
A schematic description of the method ISSH
Multi Row is shown in Figure 1. The introduction
of subsequent hashes in addition to preceding ones,
(meaning that they correspond to Q-grams generated
from overlaps of the spaced seeds located further on
the right of the current position) also implies a sig-
nificant modification of the transient phase. Specif-
ically, there are two transients, one at the beginning
and one at the end of the DNA sequence, because
the hashes “on the right” computed in the preprocess-
ing will eventually not be available, just as the hashes
“on the left” were not initially available. For these
reasons, we expect this method to perform better on
longer sequences.
3 RESULTS
Here we present the results of our experiments that
compare the newly presented Multi Spaced Seed al-
gorithms against the previously available approaches
in literature, namely FISH (Girotto et al., 2018a)
(block-based), FSH (Girotto et al., 2018b) and ISSH
(Petrucci et al., 2020) (overlap-based). All the tests
were performed consistently with the experiments
presented in previous studies. In order to evaluate
our methods under different circumstances we con-
sidered several group of spaced seeds with differ-
ent weights and lengths and computed using different
methods (maximizing the hit probability (Ounit and
Lonardi, 2016); minimizing the overlap complexity
and maximizing the sensitivity (Hahn et al., 2016)).
The spaced seeds used can be found in the appendix of
(Petrucci et al., 2020). The DNA sequences of which
Efficient Hashing of Multiple Spaced Seeds with Application
159

the hashing is computed consist in several dataset of
metagenomic reads, each with a different read count
and a different read length. All the experiments have
been performed on a laptop equipped with an Intel i9-
9980HK CPU at 2.4 GHz and 16 GB of RAM. For the
consistency of the results and comparisons presented
we run on the same machine the programs made avail-
able by the previous papers (Girotto et al., 2018b;
Girotto et al., 2018a; Petrucci et al., 2020).
The results are expressed in terms of the speedup,
that is the ratio of the time that is spent to compute the
hash using the reference method (that calculates each
hash position starting from the read in input using for-
mula (1)) over the time needed by the method that is
currently being evaluated.
3.1 Performance Evaluation
In Figure 2 is shown the speedup for the different
methods when considering the first group of 9 spaced
seeds with length 31 and weight 22. The datasets,
displayed on the x-axis, are ordered by increasing av-
erage read length.
Figure 2: Speedup of the hashing computation when con-
sidering spaced seeds W22L31.
From this graph we can see how the three new
methods presented in this paper provide a greater
speedup with respect to the previously proposed
methods. In particular we found out that, for this
group of spaced seeds, even the ISSH method reit-
erated for each of the seed obtains a higher speedup
with respect to the FISH approach.
Among all the newly proposed methods ISSH
Multi Column obtains overall the highest speedup, but
it is slightly surpassed by ISSH Multi Row for the
datasets with the longest reads. For each of the tested
methods an increase in the speedup can be noticed
when the average read length increases: this is due
to the contribution of the transient time that is more
predominant the shorter the read.
This holds especially true for ISSH Multi and
ISSH Multi Row: the first method only improves after
the transient and uses the same transient as ISSH; the
second method is additionally penalized when dealing
with short reads because it requires two transients. On
this test, these new methods can reach speedups above
17x whereas all previous methods are lower than 10x.
If we consider another group of spaced seeds, hav-
ing length 45 and weight 32 (data not shown), we can
obtain even higher speedups. Overall, on this test the
average speedup of FSH-Multi is 1.9x, FISH-Multi
7.2x, ISSH Multi Row 14.9x and ISSH Multi Column
18.1x.
Figure 3: Average speedup for all spaced seeds groups of
the hashing computation.
In Figure 3 we show the average speedup for each
group of seeds. We can see that the previous consid-
erations still hold: having groups of spaced seeds that
are longer and have a higher weight leads to higher av-
erage speedups irrespective of the method used. Even
considering the average speedup, the method rank-
ing is the same: the ISSH Multi Column approach
is the one which offers the highest speedup for each
group of spaced seeds, with ISSH Multi Row being a
close second. From Figure 2 it is possible to see how
ISSH Multi Row starts to perform better when longer
sequences are considered, even obtaining a speedup
slightly higher than ISSH Multi Column.
Our tests show how performing the hashing con-
sidering multiple spaced seeds at a time can decrease
the computation time significantly, which can be no-
ticed for each method. Using multiple spaced seeds,
the average speedup for FSH improves from 1.5x
to 1.56x, for FISH improves from 3.4x to 6.3x and
for ISSH improves from 6.8x to 13.38x, obtained by
ISSH Multi Column.
In Figure 4 we present the analysis on how
the speedup changes when changing the number of
spaced seeds considered in a group. We conducted
this test by considering the group of spaced seeds
W22L31, specifically computing the hashing using
subsets incrementally bigger up to using all the 9
spaced seeds. We can see in Figure the improvement
of using a “multi” version of the algorithm – with re-
spect to using the single seed version – when increas-
ing the number of spaced seed considered. It is inter-
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
160
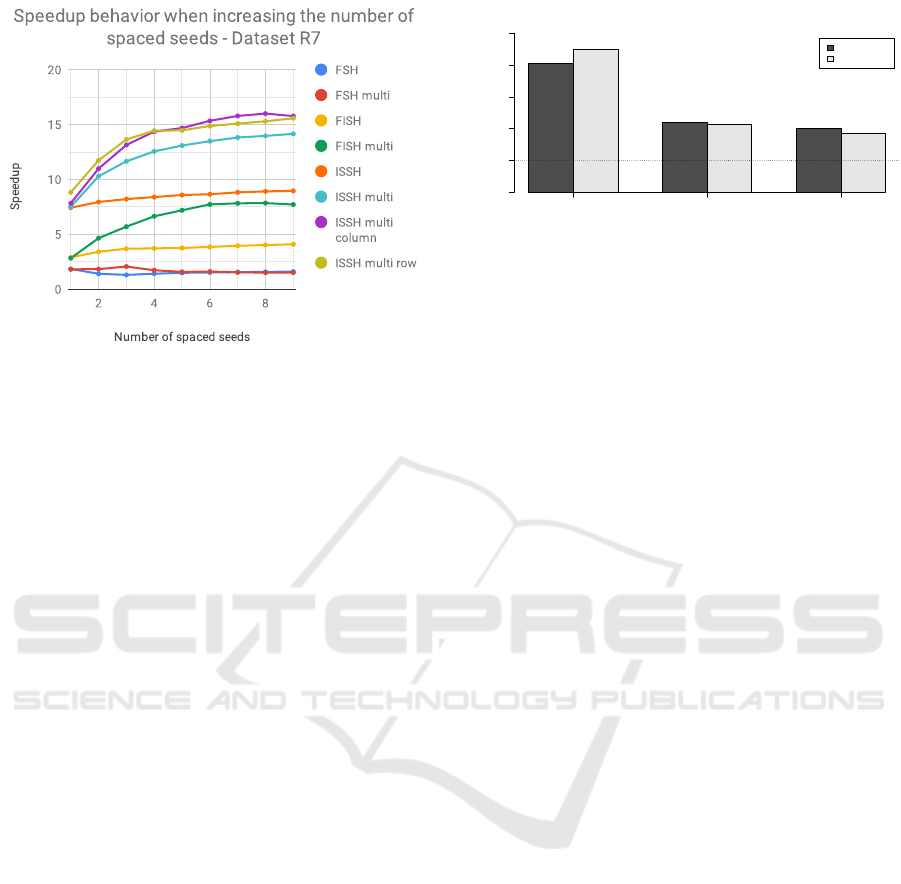
Figure 4: Speedup on dataset R7 when considering a differ-
ent number of spaced seeds.
esting to note that the highest speedup improvement
is noticeable when considering a number of spaced
seeds between 2 and 6. Obtaining comparably higher
speedups even for smaller groups of spaced seeds that
contain only 2-6 of them is an interesting result: the
proposed methods can offer significant improvements
in terms of speed even in the case of having to use less
spaced seeds for memory limitations, like with Clark-
S that uses only 3 spaced seeds (Ounit and Lonardi,
2016).
3.2 Multiple Spaced Seeds Application:
Metagenomic Classification of
Clark-S
In this section we test the ability of our hashing al-
gorithms to speed up the metagenomic classification
of reads performed by Clark-S (Ounit and Lonardi,
2016). Clark-S is the spaced seed version of Clark
and it significantly improves the sensitivity w.r.t. to
the original k-mers version. However, this gain comes
at the cost of the computational time, in fact Clark-S
is much slower than Clark. There are two main rea-
sons for this slowdown, one is the fact that Clark-S is
based on spaced seeds, the second is that it is uses not
just one, but three spaced seeds simultaneously. Thus,
the metagenomic classification of reads performed by
Clark-S is the perfect application to test our new hash-
ing algorithms.
We modified the source code of Clark-S in order
to include ISSH Multi Column, and test the speedup
with respect to the original implementation of Clark-
S. We used as test a set of short reads from the Hu-
mane Microbiome Project, SRR1804065, and a set of
long reads, named Rlong, obtained by merging the
datasets R7, R8 and R9.
Qw14 Qw18 Qw22
SRR1804065
Rlong
Speedup with Variably−Weighted Seeds
Spaced Seeds Set
speedup
0 1 2 3 4 5
4.046
4.478
2.206
2.141
2.018
1.859
Figure 5: Speedup brought by ISSH Multi Column to Clark-
S with different spaced seeds of variable weight.
In Figure 5 are reported the speedups for a set
of spaced seeds with length 32, while varying the
weight. It can be observed that, the new implemen-
tation of Clark-S is much faster than the original one,
in fact the speedup ranges between 1.85x to 4.47x. It
is worth noting that these speedups are lower than the
ones we have seen in the previous experiments. In
fact, the classification of reads performed by Clark-S
is based on the hashing values of spaced seeds, but it
also requires other specific processing and data struc-
tures. Nevertheless, we showed that efficient hashing
algorithms can be applied to Clark-S, with a signifi-
cant speedup with respect to the original implementa-
tion, and thus resolving the slowdown introduced by
the use of spaced seeds.
4 CONCLUSIONS
In this paper we present an set of new algorithms for
problem of multiple spaced seed hashing, to address
the computational slowdown that has been shown in
literature with respect to the use of k-mers.
By considering multiple spaced seeds at the same
time the methods we presented managed to exploit the
hash values already computed at preceding steps to
minimize both the number of encoding operations to
be carried out and the number of previously computed
hashes from which positions are recovered.
We reported the results on several tests, which
show that our methods offer a valuable speedup even
when considering a small number of spaced seeds,
and significantly outperform in all tests the previously
proposed algorithms, with a speedup that can reach
20x. ISSH Multi Column appears to be the fastest
algorithm, but the results reported suggest that, for
longer reads, ISSH Multi Row could offer even better
performance.
We also apply these efficient hashing algorithms
to an application in the field of metagenomic, the
classification of reads performed by Clark-S (Ounit
Efficient Hashing of Multiple Spaced Seeds with Application
161

and Lonardi, 2016), and we shown that a significant
speedup can be obtained, thus resolving the slowdown
introduced by the use of spaced seeds.
The approaches that we presented rely on a greedy
preprocessing, similarly to the one adopted by ISSH.
Possible future extensions could focus on improving
this preprocessing: instead of using a greedy pol-
icy for selecting the group of previously computed
hashes from which to extract the positions to reuse,
it could be beneficial to investigate global optimiza-
tion schemes in order to make the computation even
faster.
REFERENCES
Apostolico, A., Guerra, C., Landau, G. M., and Pizzi,
C. (2016). Sequence similarity measures based on
bounded hamming distance. Theoretical Computer
Science, 638:76 – 90.
A.Zielezinski, Vinga, S., Almeida, J., and et al. (2017).
Alignment-free sequence comparison: benefits, appli-
cations, and tools. Genome Biol, 18:186.
B
ˇ
rinda, K., Sykulski, M., and Kucherov, G. (2015). Spaced
seeds improve k-mer-based metagenomic classifica-
tion. Bioinformatics, 31(22):3584.
Dencker, T., Leimeister, C.-A., Gerth, M., Bleidorn, C.,
Snir, S., and Morgenstern, B. (2019). ‘Multi-SpaM’:
a maximum-likelihood approach to phylogeny recon-
struction using multiple spaced-word matches and
quartet trees. NAR Genomics and Bioinformatics,
2(1). lqz013.
Girotto, S., Comin, M., and Pizzi, C. (2017a). Fast spaced
seed hashing. In Proceedings of the 17th Workshop
on Algorithms in Bioinformatics (WABI), volume 88
of Leibniz International Proceedings in Informatics,
pages 7:1–7:14.
Girotto, S., Comin, M., and Pizzi, C. (2017b). Metage-
nomic reads binning with spaced seeds. Theoretical
Computer Science, 698:88–99.
Girotto, S., Comin, M., and Pizzi, C. (2018a). Efficient
computation of spaced seed hashing with block index-
ing. BMC Bioinformatics, 19(15):441.
Girotto, S., Comin, M., and Pizzi, C. (2018b). Fsh: fast
spaced seed hashing exploiting adjacent hashes. Al-
gorithms for Molecular Biology, 13(1):8.
Hahn, L., Leimeister, C.-A., Ounit, R., Lonardi, S.,
and Morgenstern, B. (2016). Rasbhari: Optimiz-
ing spaced seeds for database searching, read map-
ping and alignment-free sequence comparison. PLOS
Computational Biology, 12(10):1–18.
Harris, R. S. (2007). Improved Pairwise Alignment of Ge-
nomic Dna. PhD thesis, University Park, PA, USA.
Kucherov, G., No
´
e, L., and Roytberg, M. A. (2006). A uni-
fying framework for seed sensitivity and its applica-
tion to subset seeds. Journal of Bioinformatics and
Computational Biology, 4(2):553–569.
Leimeister, C. and Morgenstern, B. (2014). Kmacs: the
k-mismatch average common substring approach to
alignment-free sequence comparison. Bioinformat-
ics., 30(14):2000–8.
Leimeister, C.-A., Boden, M., Horwege, S., Lindner, S., and
Morgenstern, B. (2014). Fast alignment-free sequence
comparison using spaced-word frequencies. Bioinfor-
matics, 30(14):1991.
Li, Y. and Ilie, L. (2017). Sprint: ultrafast protein–protein
interaction prediction of the entire human interac-
tome. BMC Bioinformatics, 18(485).
Ma, B., Tromp, J., and Li, M. (2002). Patternhunter: faster
and more sensitive homology search. Bioinformatics,
18(3):440.
Marc¸ais, G., Solomon, B., Patro, R., and Kingsford, C.
(2019). Sketching and sublinear data structures in ge-
nomics. Annual Review of Biomedical Data Science,
2(1):93–118.
Mohamadi, H., Chu, J., Vandervalk, B. P., and Birol, I.
(2016). ntHash: recursive nucleotide hashing. Bioin-
formatics, page btw397.
No
´
e, L. and Martin, D. E. K. (2014). A coverage criterion
for spaced seeds and its applications to support vector
machine string kernels and k-mer distances. Journal
of Computational Biology, 21(12):947–963.
Onodera, T. and Shibuya, T. (2013). The gapped spectrum
kernel for support vector machines. In Proceedings
of the 9th Conference on Machine Learning and Data
Mining in Pattern Recognition, MLDM’13, pages 1–
15. Springer-Verlag.
Ounit, R. and Lonardi, S. (2016). Higher classification
sensitivity of short metagenomic reads with clark-s.
Bioinformatics, 32(24):3823.
Ounit, R., Wanamaker, S., Close, T. J., and Lonardi, S.
(2015). Clark: fast and accurate classification of
metagenomic and genomic sequences using discrim-
inative k-mers. BMC Genomics, 16(1):1–13.
Petrucci, E., No
´
e, L., Pizzi, C., and Comin, M. (2020). It-
erative spaced seed hashing: Closing the gap between
spaced seed hashing and k-mer hashing. Journal of
Computational Biology, 27(2):223–233.
Rumble, S. M., Lacroute, P., Dalca, A. V., Fiume, M.,
Sidow, A., and Brudno, M. (2009). Shrimp: Accurate
mapping of short color-space reads. PLOS Computa-
tional Biology, 5(5):1–11.
R
¨
ohling, S., Linne, A., Schellhorn, J., Hosseini, M.,
Dencker, T., and Morgenstern, B. (2020). The num-
ber of k-mer matches between two dna sequences as
a function of k and applications to estimate phyloge-
netic distances. PLoS One, 15.
Wood, D., Lu, J., and Langmead, B. (2019). Improved
metagenomic analysis with kraken 2. Genome Biol,
20(257).
Wood, D. E. and Salzberg, S. L. (2014). Kraken: ultra-
fast metagenomic sequence classification using exact
alignments. Genome Biology, 15:R46.
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
162
