
Towards Computer Assisted Compliance Assessment in the Development
of Software as a Medical Device
Sadra Farshid
1,2 a
, Bruno Lima
1,3 b
and Jo
˜
ao Pascoal Faria
1,3 c
1
Faculty of Engineering of the University of Porto, Porto, Portugal
2
Capgemini Engineering Portugal, Lisbon, Portugal
3
INESC TEC, Porto, Portugal
Keywords:
Medical Device, Software as a Medical Device, Compliance Assessment, Web Platform.
Abstract:
Medical devices (MDs) and Software as a Medical Device (SaMD) are essential for e-Health applications,
but they must comply with strict standards and regulations to ensure their safety and effectiveness. However,
there is a lack of tools to assist in conducting appraisals for compliance assessment and managing appraisal
information. In this paper, after reviewing the most relevant standards and regulations for MD and SaMD cer-
tification, we propose a web platform to help technology companies that lack expertise in developing SaMD to
create compliant and high-quality products for the e-Health market. The platform provides users with custom
checklists or questionnaires depending on the selected regulations, standards, risk classes, and product pa-
rameters. Supporting a secure, incremental, and collaborative approach to completing the assessment process,
the platform enables the attachment of notes, evidence, and improvement suggestions. It facilitates repeated
assessments over time for data reuse and comparative analysis, enhancing the assessment process’s efficiency
and effectiveness.
1 INTRODUCTION
Numerous companies are engaged in the develop-
ment of various types of software applications. How-
ever, when they enter the SaMD market, they often
lack sufficient knowledge of the standards and regu-
lations specific to SaMD. Various local and interna-
tional rules and regulations control the medical de-
vice (MD) market. In order for an MD to be sold, it
must comply with these regulations. The regulations
are complex and strict, making it difficult for small
and medium-sized enterprises (SMEs) to gain medi-
cal approval for their products.
To meet this challenge, we propose in this pa-
per a web platform to help MD companies achieve
compliance for their medical products. The platform
will support the implementation of best practices for
the development, testing, and validation of MDs and
SaMD in the e-Health domain, taking into account
regulatory requirements outlined in standards such as
IEC 62304 (Jordan, 2006). The goal of this proposal
a
https://orcid.org/0009-0004-3705-6259
b
https://orcid.org/0000-0003-2572-047X
c
https://orcid.org/0000-0003-3825-3954
is to create a tool that will help ensure compliance in
the development of MDs, specifically software as a
medical device (SaMD) (Group, 2019).
The remaining sections of the paper are structured
as follows. A brief characterization of the different
types of MDs is presented in Section 2. An overview
of the standards and regulations for MD and SaMD
certification is presented in section 3. Section 4 pro-
vides a brief overview of existing compliance assess-
ment tools. A description of the proposed web plat-
form for compliance assessment is provided in section
5. Lastly, section 6 concludes the paper.
2 TYPES OF MDs
An MD can be defined as any instrument, device, soft-
ware, implant, material, or other article that is in-
tended by the manufacturer to be used for medical
purposes in or on the human body (Teferra, 2017).
These purposes may include the diagnosis, preven-
tion, monitoring, prediction, prognosis, treatment,
or alleviation of disease; the diagnosis, monitoring,
treatment, alleviation, or compensation for injury or
disability; the investigation, replacement, or modifi-
728
Farshid, S., Lima, B. and Faria, J.
Towards Computer Assisted Compliance Assessment in the Development of Software as a Medical Device.
DOI: 10.5220/0012148400003538
In Proceedings of the 18th International Conference on Software Technologies (ICSOFT 2023), pages 728-735
ISBN: 978-989-758-665-1; ISSN: 2184-2833
Copyright
c
2023 by SCITEPRESS – Science and Technology Publications, Lda. Under CC license (CC BY-NC-ND 4.0)

cation of the anatomy or physiology; or the provision
of information through the examination of specimens.
An Active Medical Device (AMD)(Group, 2019)
(IMDRF, 2018) is a type of MD that uses energy to
function. AMDs are typically used to diagnose, pre-
vent, monitor, or treat a medical condition or dis-
ease. AMDs can be either invasive (meaning they
involve some type of penetration into the body) or
non-invasive (meaning they do not come into direct
contact with the body). Some examples of AMDs in-
clude ultrasound machines, x-ray machines, and laser
surgery devices. AMDs are regulated by authorities
such as the US Food and Drug Administration (FDA)
and the European Medicines Agency (EMA) to en-
sure their safety and effectiveness.
An In Vitro Diagnostic Medical Device (IVD)
(Group, 2019) is a type of MD used to perform di-
agnostic tests on samples of bodily fluids or tissues
taken from the human body in order to detect diseases,
infections, or other conditions. These devices are reg-
ulated by government agencies to ensure their accu-
racy and safety and must comply with the regulations
and standards.
Software as Medical Device (SaMD) (Group,
2019) is a type of software that is intended to be used
for one or more medical purposes, such as diagno-
sis, prevention, monitoring, or treatment of a medical
condition or disease. SaMD is classified as an MD
and includes IVDs. It can be run on general-purpose
computing platforms and does not need to be part of a
hardware MD in order to perform its intended medi-
cal functions. However, if the software’s primary pur-
pose is to drive a hardware MD, it is not considered
SaMD. SaMD may be used in combination with other
products, including MDs, and may be interfaced with
other MDs and software. Besides generic regulations
for MDs, SaMD is also subject to specific regulations.
3 REGULATIONS FOR MD AND
SaMD CERTIFICATION
MDs and SaMD must follow standards that are set
by the International Organization for Standardization
(ISO) (Heires, 2008) (iso, ), the International Elec-
trotechnical Commission (IEC) (iec, b), the European
Union regulation bodies (Kramer et al., 2012), the
Food and Drug Administration (FDA)., and the Inter-
national Medical Device Regulators Forum (IMDRF)
(Group et al., 2020). These standards are used to en-
sure the safety and effectiveness of these products.
The International Medical Device Regulators Fo-
rum (IMDRF) (Group et al., 2020) is a global or-
ganization that brings together MD regulators from
around the world to collaborate on the development
of international guidelines and standards for the regu-
lation of MDs. The IMDRF was founded in 2011 as
a successor to the Global Harmonization Task Force
on Medical Devices (GHTF) (Gagliardi, 2009). The
organization aims to promote the safety, quality, and
performance of MDs by facilitating the development
of internationally recognized standards and guidelines
for the regulation of MDs.
The applicable standards and regulations for
SaMD and MDs are described next.
3.1 EU MDR
The European Union Medical Device Regulation (EU
MDR) (Kramer et al., 2012) sets out the rules for
the design, production, and performance of MDs in
the EU. It replaces the Medical Devices Directive
(MDD), which had been in place since 1993. The
MDR aims to improve the safety and performance of
MDs and to increase transparency and accountability
in the MD market.
The EU MDR applies to all MDs that are placed
on the EU market, regardless of where they are manu-
factured. It covers a wide range of products, including
everything from simple bandages and tongue depres-
sors to complex diagnostic and therapeutic devices
such as pacemakers and MRI machines.
The EU MDR requires MD manufacturers to
demonstrate the safety and effectiveness of their prod-
ucts through clinical data and other evidence. It also
establishes a new regulatory framework for innova-
tive MDs, including those that use software or rely on
digital technologies.
It includes 17 annexes that cover different aspects
of MDs (med, ), such as: general safety and per-
formance requirements; essential principles of safety
and performance; clinical evaluation; classification
of MDs; conformity assessment procedures; marking
and labeling of MDs; vigilance and market surveil-
lance; registration of manufacturers, authorized rep-
resentatives, and importers; clinical investigations;
transitional provisions; specific requirements for IV-
DRs; specific rules on the safety and performance
of custom-made and investigational MDs, active im-
plantable medical devices, MDs meant to administer
medicinal products, MDs meant to be used in contact
with blood, body fluids or tissues, MDs meant to be
used for dental purposes, and MDs meant to be used
for human reproduction.
To help determine if a given product is in the
scope of the EU MDR, specific guidance is provided
in (Group, 2019), in the form of a decision procedure
schematized in Fig. 1.
Towards Computer Assisted Compliance Assessment in the Development of Software as a Medical Device
729
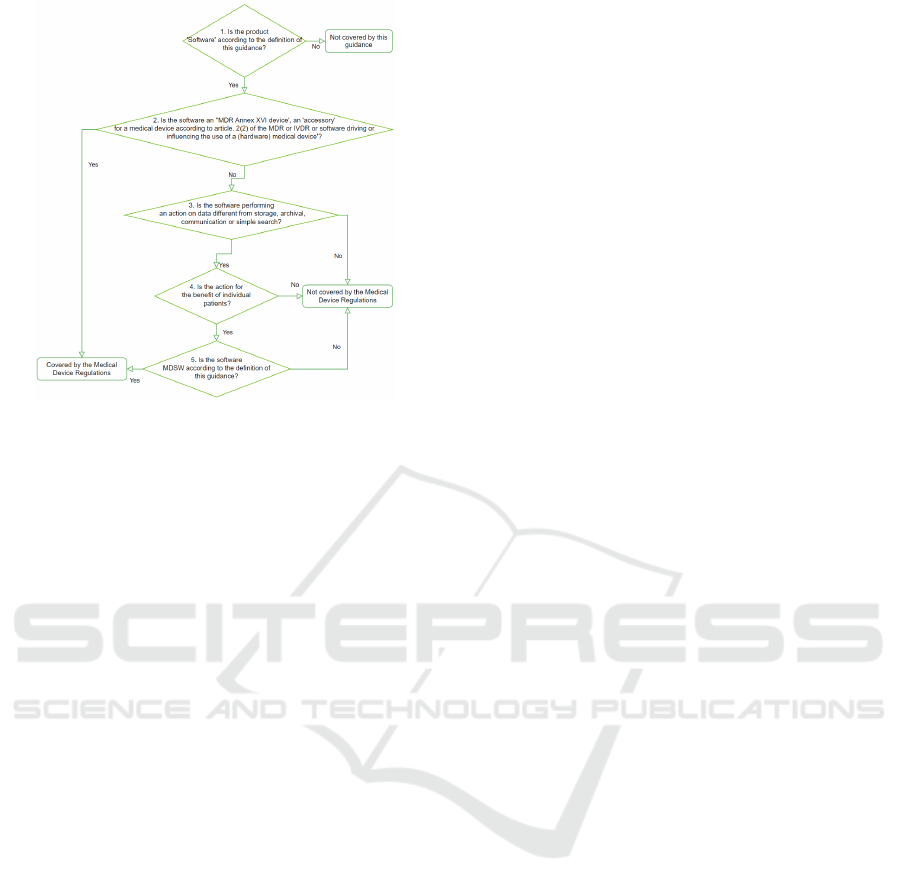
Figure 1: Decision steps to assist qualification of SaMD
according to (Group, 2019).
3.2 FDA Regulations
FDA is a federal agency within the United States De-
partment of Health and Human Services (fda, ). It is
responsible for protecting public health by ensuring
the safety, efficacy, and security of human and veteri-
nary drugs, biological products, and MDs.
The FDA’s regulation of these products covers the
following areas:
• Premarket Review and Clearance/Approval -
Before a drug, biological product, or MD can be
marketed in the U.S., it must go through a pre-
market review process to ensure that it is safe and
effective. The FDA will clear or approve the prod-
uct depending on the level of risk it poses to pa-
tients.
• Postmarket Surveillance - After a product is on
the market, the FDA will continue to monitor its
safety and efficacy through postmarket surveil-
lance. This includes monitoring reports of adverse
events and taking action if necessary.
• Manufacturing and Quality Control - The FDA
sets standards for the manufacturing and quality
control of drugs, biological products, and MDs to
ensure that they meet certain quality standards.
• Labeling and Advertising - The FDA regulates
the labeling and advertising of drugs, biological
products, and MDs to ensure that they are truthful
and not misleading.
3.3 IEC 62304
IEC 62304 is an international standard that provides
guidance on the development of medical software. It
covers the entire lifecycle of medical software, in-
cluding design, development, testing, maintenance,
and decommissioning. The standard is intended to
help organizations ensure that their medical software
is safe and effective for use in the healthcare environ-
ment.
The standard consists of a number of different sec-
tions, each of which covers a specific aspect of medi-
cal software development.
The standard mandates organizations to identify
and evaluate the risks associated with their medical
software, and put in place appropriate controls to
manage these risks. The software systems are clas-
sified into three categories, Class A, Class B, or Class
C, based on their potential impact on patient safety
(see Fig. 2). Class A systems pose a minimal risk,
Class B pose a moderate risk, and Class C poses a
high risk to patients.
The standard also provides guidance on how to
plan, design, implement, test, and maintain medical
software, and the use of configuration management
and change control processes. It also requires organi-
zations to verify and validate their medical software
at various stages of the development process and pro-
vides guidance on how to maintain and decommission
the software when it is no longer needed, including
the handling of data and disposal of hardware.
3.4 ISO 14971
ISO 14971 (Teferra, 2017) is an international stan-
dard that provides guidance on the application of risk
management to the design and development of MDs.
It outlines a systematic approach for identifying, eval-
uating, and mitigating risks associated with the use of
MDs. The standard is intended to help manufacturers
ensure that their products are safe and effective for
their intended use, and to provide a common frame-
work for regulators to evaluate the risk management
processes of MD manufacturers.
According to ISO 14971, risks associated with
MDs can be classified into several categories, includ-
ing:
• Physical Risks - which relate to the physical
properties of the device or its components, and
may include risks such as electrical shock, me-
chanical failure, or chemical exposure.
• Performance Risks - which relate to the device’s
ability to function as intended, and may include
ICSOFT 2023 - 18th International Conference on Software Technologies
730
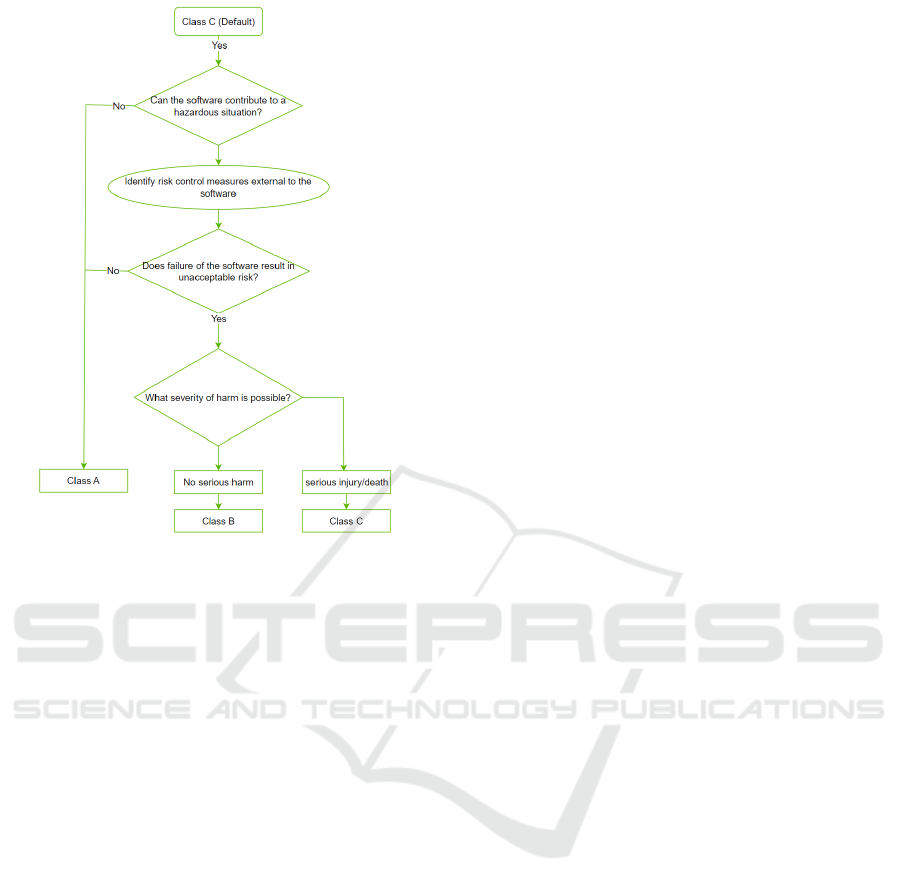
Figure 2: Risk classification according to IEC 62304.
risks such as incorrect diagnosis, incorrect treat-
ment, or inadequate performance.
• Use-Related Risks - which relate to the way in
which the device is used, and may include risks
such as incorrect handling, incorrect maintenance,
or incorrect disposal.
• Human Factors Risks - which relate to the inter-
action between the device and its users, and may
include risks such as user error, user fatigue, or
user confusion.
• Environmental Risks - which relate to the exter-
nal environment in which the device is used, and
may include risks such as extreme temperatures,
humidity, or radiation.
There are several ways to classify risks based on
their likelihood and severity. One common approach
is to use a matrix or grid to plot the likelihood and
severity of risks, with the resulting quadrants indicat-
ing the overall level of risk. For example, a risk might
be classified as high likelihood and high severity (a
high-risk situation), low likelihood and low severity
(a low-risk situation), or anywhere in between.
3.5 ISO 13485
ISO 13485 is an international standard that speci-
fies requirements for a quality management system
(QMS) in the design, development, production, instal-
lation and servicing of MDs. The standard is intended
to help organizations in the MD industry meet the ap-
plicable regulatory requirements and to demonstrate
their ability to provide MDs and related services that
consistently meet customer and applicable regulatory
requirements.
The standard is based on the ISO 9001:2015 stan-
dard for quality management systems, with additional
requirements specific to the MD industry. These in-
clude requirements for risk management, design and
development, production, installation, and servicing,
as well as regulatory and legal compliance.
The ISO 13485 standard is widely recognized
and used by MD manufacturers and other organiza-
tions around the world as a means of demonstrat-
ing their commitment to quality and safety in the de-
sign, production, and servicing of MDs. It is also of-
ten required by regulatory authorities as a means of
demonstrating compliance with relevant regulations
and standards (Bos, 2018).
This standard requires the organization to meet a
number of requirements specified in it. Organizations
must establish, document, implement, maintain, and
continuously improve a Quality Management System
(QMS) that meets the requirements of the standard.
Senior management must demonstrate their commit-
ment to the development and implementation of the
QMS and to the continuous improvement of the or-
ganization’s products and processes. The organiza-
tion must provide the necessary resources to imple-
ment and maintain the QMS, including personnel, in-
frastructure, and work environment. The organization
must plan, develop, produce, and deliver MDs that
meet customer and regulatory requirements. They
must monitor, measure, and analyze its processes
and products to identify opportunities for improve-
ment and take appropriate corrective and preventive
actions. The organization must conduct internal au-
dits to verify that the QMS is being effectively imple-
mented and maintained, and senior management must
review the QMS at defined intervals to ensure its on-
going suitability, adequacy, and effectiveness.
3.6 In Vitro Diagnostic Regulation
(IVDR)
The In Vitro Diagnostic Regulation (IVDR) is a Euro-
pean Union (EU) regulation that sets out the require-
ments for the design, production, and performance
evaluation of IVD. The IVDR applies to all IVDs that
are used for the examination of specimens, includ-
ing blood, tissue, and other substances, taken from
the human body for the purpose of providing infor-
mation for the diagnosis, prevention, monitoring, or
Towards Computer Assisted Compliance Assessment in the Development of Software as a Medical Device
731

Figure 3: Appraiser Assistant.
prediction of a disease or condition. The regulation
aims to ensure the safety, performance, and effective-
ness of IVDs, as well as to provide a consistent and
harmonized regulatory framework for their marketing
and use in the EU. The IVDR replaces the previous
In Vitro Diagnostic Directive (IVDD) and came into
force on May 26, 2022. Some aspects covered by the
IVDR include: risk classification of IVDs; confor-
mity assessment procedures; clinical evidence; label-
ing and instructions for use; post-market surveillance;
registration and listing of IVDs.
In addition to the above standards and regulations,
MDs may also be subject to a variety of other national
and regional regulations, depending on the country or
region in which they are sold(Group, 2019).
4 COMPLIANCE ASSESSMENT
RESOURCES
Currently, the only resources available for evaluating
e-Health and SaMD products are checklists and desk-
top applications in related domains, such as Appraisal
Assistant (app, ) for CMMI (Chrissis et al., 2011). An
example screen of this application is demonstrated in
Fig. 3. However, these tools can be difficult for SMEs
to use effectively when assessing SaMD products, as
they must meet strict regulations. The lack of a web
platform specifically designed for the e-Health and
SaMD domain adds to the challenges faced by these
businesses in the product assessment process.
In addition to the checklists and desktop applica-
tions mentioned earlier, some websites are available
at the time this paper was written that offer consult-
ing services to assist manufacturers of MD and SaMD
devices with the assessment process. These websites
served as an alternative option for companies seek-
ing guidance and support with the assessment of their
devices. To our knowledge, there are currently no
web-based platforms available for the evaluation of
e-Health and SaMD products.
Some commercial quality management software
solutions are also available to aid organizations in
the life sciences and medical device sectors in es-
tablishing quality management systems that enhance
compliance with multiple regulations and standards.
However, these solutions are generally not tailored for
compliance assessments conducted by manufacturers
or third-party entities. Our platform aims to fill this
niche - it is designed specifically to facilitate thorough
compliance assessments, setting it apart from conven-
tional quality management tools.
5 PROPOSED COMPLIANCE
ASSESSMENT PLATFORM
In order to facilitate the compliance assessment of
MD and SaMD and the management of assessment
information, we propose a web platform with the
functionalities summarized in Fig. 4 and described
next.
The main goals of the platform are to:
• provide to the user (appraiser) the applicable as-
sessment checklist or questionnaire, depending on
the selected regulation or standard, risk class, and
other relevant parameters of the product under as-
sessment;
• guide the users in conducting risk assessment and
classification of a product under assessment based
on relevant decision trees and checklists;
• provide an easily accessible platform for users to
answer assessment checklists or questionnaires in
a collaborative, secure, and incremental way;
• provide a way for users to attach explanatory
notes and evidence to each item or answer so that
relevant stakeholders can review them (including
auditors in a formal certification process);
• support review and approval workflows;
• provide a way for users to attach improvement
proposals to overcome issues identified;
• provide an easy way for users to visualize the
overall results of finished assessments;
• allow users to export the assessment results and
data to reports generated according to previously
defined templates;
• allow users to conduct multiple assessments of
the same product over time, possibly reusing data
from previous assessments, and easily comparing
their results.
ICSOFT 2023 - 18th International Conference on Software Technologies
732
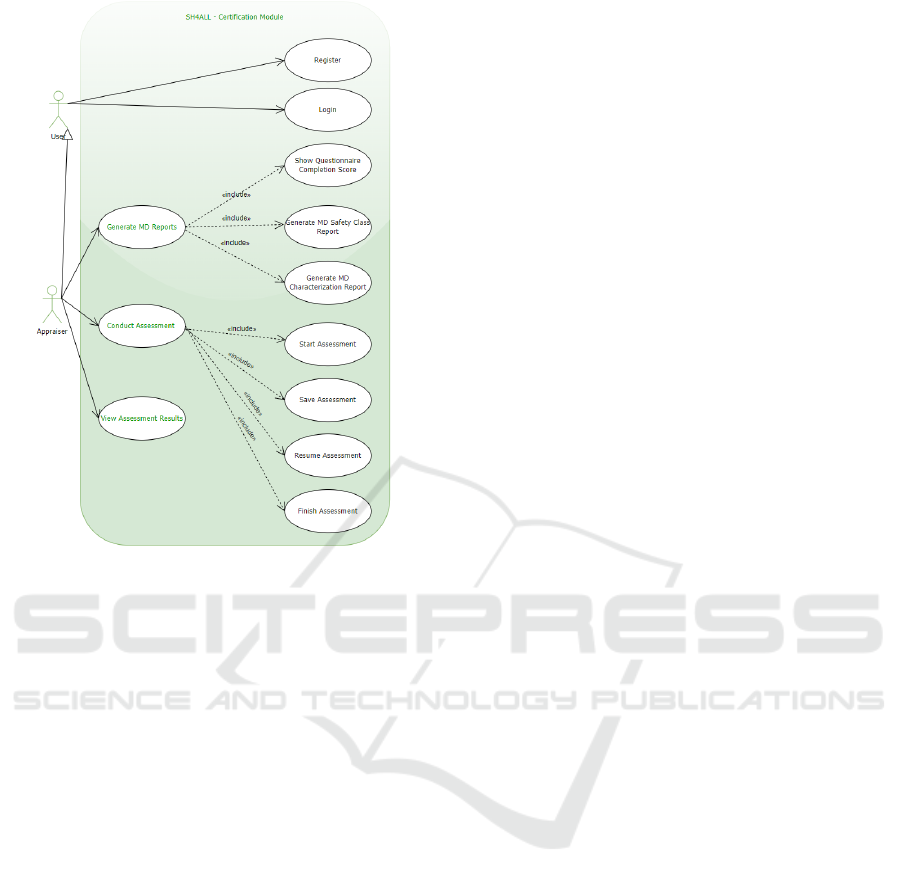
Figure 4: Use Case Diagram of the Proposed Web Platform.
5.1 Functionalities
The users (appraisers) can register and log in to the
web platform, where they can conduct assessments
after being authenticated. The assessment process can
be saved and resumed by the user at a later time, and
the module also has an auto-saving function that sub-
mits the answers to the questionnaire to the server and
saves them in the backend data store. Upon comple-
tion of the assessment, the module generates the as-
sessment result and characterizes the MD in a report.
It also provides scores for the completion of the total
questionnaire and the completion level of each sec-
tion.
More specifically, the main functionalities pro-
vided to the user are:
1. Register - In order to be able to access the func-
tionalities provided by the web platform, the user
has first to register on the website, with appropri-
ate credentials.
2. Login - Once registered in the web platform, the
user can log in to the web platform and access the
functionalities provided next.
3. Conduct Assessment - The appraiser can con-
duct an assessment by answering the questions of
a questionnaire, dependent on a risk classification
of the MD or SaMD under analysis.
4. Start Assessment - This use case is the first step
in conducting an assessment. Appraiser starts an-
swering the question.
5. Save Assessment - Since an assessment is rarely
concluded in a single session, the user can save
the assessment information defined so far.
6. Resume Assessment - The user can resume the
assessment process.
7. Finish Assessment - An appraiser can conclude
the assessment process, after answering the ques-
tionnaire provided by the system.
8. Generate MD Report - After concluding the
questionnaire, multiple MD reports can be gen-
erated.
9. View Assessment Result - The user can see the
assessment result.
10. Show Questionnaire Completion Score - After
concluding a questionnaire, a score is generated
indicating the completion level of the question-
naire.
11. Generate MD Safety Class Report - This report
identifies the safety class of the MD, according to
IEC 62304.
In conducting an assessment, the platform offers
various features. Initially, only one standard is con-
sidered, and it can be extended to support more stan-
dards and regulations in the future as per require-
ments. The process of conducting an assessment be-
gins by choosing a standard from a list of standards,
which is illustrated in Fig. 5. The platform also sup-
ports multi-session assessment, enabling the appraiser
to save progress and continue later. Additionally, mul-
tiple assessments for one device are supported. This
enables the appraiser to assess their device based on
various standards and regulations. On the platform
dashboard, a list of the assessments and a summary
of the assessments appears. Once the risk level of the
MD under analysis is determined, the system presents
a checklist with a series of questions for the appraiser
to answer. These questions cover sections 4 to 9 of
IEC 62304 (iec, a), illustrated in Fig. 6. To ensure the
accuracy and completeness of the assessment, the ap-
praiser can provide supportive evidence for each an-
swer. If a question is not applicable, the appraiser can
mark it with a comment explaining the reason. After
the assessment is completed, the score is computed
for each section and the entire assessment and then
displayed to the appraiser. The score is shown to the
appraisar as it is illustrated in Fig. 7.
Towards Computer Assisted Compliance Assessment in the Development of Software as a Medical Device
733

Figure 5: Certification Assessment Assistant, Standard Se-
lection page.
Figure 6: Certification Assessment Assistant, Questionnaire
page.
Figure 7: Certification Assessment Assistant, Score Result
page.
5.2 Architecture
5.2.1 Frontend
The frontend will be built on Angular
1
, an efficient
framework for creating sophisticated single-page ap-
plications. Splitting a web portal into smaller Angular
applications enables agile development and mainte-
nance, independent deployment, and scalable server
1
https://angular.io/guide/architecture/
management.
5.2.2 Backend
To make business capabilities and functionalities ac-
cessible to the clients such as mobile and web portals,
we will create small RESTful (Richardson and Ruby,
2008) services. These services will be developed us-
ing Kotlin (Jemerov and Isakova, 2017) in combina-
tion with Springboot (Walls, 2015) (spr, ). By using
Kotlin and Springboot, we can create lightweight ser-
vices that clients can easily consume.
Kotlin is a programming language that combines
object-oriented and functional programming concepts
and is designed to be interoperable, safe, clear, and
well-supported by tools. It was originally created for
use with the Java Virtual Machine (JVM) (Yellin and
Lindholm, 1996) and Android, but can also be used
to create applications for JavaScript and native code.
Kotlin was developed by JetBrains, the company be-
hind the IntelliJ IDEA (Krochmalski, 2014)
2
devel-
opment environment, and has been open source since
2012.
6 CONCLUSION AND FUTURE
WORK
Before MDs can be released to the market, they must
comply with various standards and regulations to en-
sure their safety and effectiveness. To do this, it is im-
portant to identify and assess the potential risks asso-
ciated with the MD during the development process.
Multiple organizations such as ISO, IEC, and
FDA provide guidelines and standards for MD devel-
opment and certification. MDs are classified accord-
ing to their usage, life cycle, and risk level, and manu-
facturers must certify their devices according to these
guidelines.
A proposal for a web platform is made to help
manufacturers ensure their devices meet the regula-
tions. The initial step in developing an MD is to en-
sure that it meets the necessary qualifications. This
is done by having an appraiser conduct a question-
naire based on the risk class of the device. The ques-
tionnaire is designed to evaluate the device’s compli-
ance with the relevant standards and regulations, and
it serves as the first deliverable software product in
the development process. The results of this question-
naire will be used to determine if the device meets the
necessary qualifications and can proceed to the next
stages of development.
2
https://www.jetbrains.com/idea/
ICSOFT 2023 - 18th International Conference on Software Technologies
734

In future work, a decision tree will be imple-
mented to aid in the classification of the risk asso-
ciated with the MD. This decision tree will be used
as a tool to assist in the identification and assessment
of potential risks during the development process and
to ensure that the device meets the necessary qualifi-
cations and complies with the relevant standards and
regulations. Additionally, expanding the platform to
support additional standards and regulations in the fu-
ture, depending on the requirements, is possible.
ACKNOWLEDGMENTS
This work is a result of project SMART-HEALTH-4-
ALL - Smart medical technologies for better health
and care, with reference POCI-01-0247-FEDER-
046115, co-funded by the European Regional Devel-
opment Fund (ERDF), through the Operational Pro-
gramme for Competitiveness and Internationalization
(COMPETE 2020) and the Lisbon Regional Opera-
tional Programme (LISBOA 2020), under the POR-
TUGAL 2020 Partnership Agreement.
REFERENCES
Appraisal Assistant Beta - Official Website. https://www.
jetbrains.com/idea/features/. Accessed: 2023-01-12.
FDA - Official Website. https://www.fda.gov/. Accessed:
2023-01-12.
IEC. https://www.iso.org/. Accessed: 2023-01-12.
IEC - Official Website. https://iec.ch/. Accessed: 2023-01-
12.
ISO - Official Website. https://www.iso.org/. Accessed:
2023-01-11.
medical device regulation - Official Website. https://www.
medical-device-regulation.eu/mdr-annexes/. Ac-
cessed: 2023-01-12.
Springboot Overview - Official Website. https://spring.io/
projects/spring-boot/. Accessed: 2023-01-12.
Bos, G. (2018). Iso 13485: 2003/2016—medical de-
vices—quality management systems—requirements
for regulatory purposes. In Handbook of Medical De-
vice Regulatory Affairs in Asia.
Chrissis, M. B., Konrad, M., and Shrum, S. (2011). CMMI
for development: guidelines for process integration
and product improvement. Pearson Education.
Gagliardi, J. (2009). The global harmonization task force:
what you need to know. Biomedical Instrumentation
& Technology.
Group, M. D. C. (2019). Guidance on qualification and clas-
sification of software in regulation (eu) 2017/745–mdr
and regulation (eu) 2017/746–ivdr.
Group, M. D. C. W. et al. (2020). Principles and prac-
tices for medical device cybersecurity. In Interna-
tional Medical Device Regulators Forum.
Heires, M. (2008). The international organization for stan-
dardization (iso). New Political Economy.
IMDRF (2018). International medical device regulators fo-
rum.
Jemerov, D. and Isakova, S. (2017). Kotlin in action. Simon
and Schuster.
Jordan, P. (2006). Standard iec 62304-medical device
software-software lifecycle processes.
Kramer, D. B., Xu, S., and Kesselheim, A. S. (2012). Reg-
ulation of medical devices in the united states and eu-
ropean union.
Krochmalski, J. (2014). IntelliJ IDEA Essentials. Packt
Publishing Ltd.
Richardson, L. and Ruby, S. (2008). RESTful web services.
Teferra, M. N. (2017). Iso 14971-medical device risk man-
agement standard. International Journal of Latest Re-
search in Engineering and Technology (IJLRET).
Walls, C. (2015). Spring Boot in action. Simon and Schus-
ter.
Yellin, F. and Lindholm, T. (1996). The java virtual machine
specification.
Towards Computer Assisted Compliance Assessment in the Development of Software as a Medical Device
735
