
Random Neural Network Ensemble for Very High Dimensional Datasets
Jesus S. Aguilar–Ruiz
1 a
and Matteo Fratini
2 b
1
Pablo de Olavide University, ES–41013, Spain
2
Pharmagest, Italy
Keywords:
Neural Network, Ensemble, Random Forest, Very High Dimensionality.
Abstract:
This paper introduces a machine learning method, Neural Network Ensemble (NNE), which combines en-
semble learning principles with neural networks for classification tasks, particularly in the context of gene
expression analysis. While the concept of weak learnability equalling strong learnability has been previ-
ously discussed, NNE’s unique features, such as addressing high dimensionality and blending Random Forest
principles with experimental parameters, distinguish it within the ensemble landscape. The study evaluates
NNE’s performance across five very high dimensional datasets, demonstrating competitive results compared
to benchmark methods. Further analysis of the ensemble configuration, with respect to using variable–size
neural networks units and guiding the selection of input variables would improve the classification perfor-
mance of NNE–based architectures.
1 INTRODUCTION
Classifying patient conditions, whether binary or
multiple, through machine learning (ML) algorithms
has long been a focal point in both medicine and
bioinformatics. This paradigm finds significant ap-
plication in the analysis of gene expression profiles,
which represent the differential expression of genes in
individuals, often obtained through sequencing tech-
niques such as RNA–Seq. Differences in gene expres-
sion levels typically signify alterations in the cellu-
lar, tissue, or even organismal states. By analyzing
these profiles, characteristic patterns for various dis-
eases can be identified and annotated based on the ob-
served under or over–expression of genes compared
to a reference, which is conventionally established.
Cancer gene expression data is notably complex
due to the prevalence of single nucleotide poly-
morphism (SNP) mutations occurring throughout the
genome. This results in a correspondingly large num-
ber of features (dimensions) to consider, often encom-
passing the most representative genes associated with
the disease. Managing this type of datasets with tens
of thousands of dimensions can encounter the curse
of dimensionality, a concept originally articulated in
(Bellman, 1961), which underscores the challenge of
uncovering latent structures in datasets with a high
a
https://orcid.org/0000-0002-2666-293X
b
https://orcid.org/0009-0002-0034-9694
variability of variables. As the number of explanatory
variables increases, so does the complexity of iden-
tifying these structures, particularly evident in tasks
like feature selection for model fitting (Trunk, 1979).
The curse of dimensionality encapsulates the ex-
ponential increase in complexity, especially pro-
nounced in intricate problems with numerous vari-
ables, rendering dimensionality overwhelmingly dif-
ficult to manage (Wellinger and Aguilar-Ruiz, 2022).
Therefore, it becomes imperative to employ tech-
niques for dimensionality reduction, ensuring that in-
formation loss is minimized in the process.
Over the past two decades, ML approaches have
offered a robust framework for resolving gene ex-
pression classification problems (Hwang et al., 2002;
Deng et al., 2019). With a diverse range of method-
ologies available, new techniques called Ensembles
have emerged, combining existing methods and of-
ten proving more successful than individual ones (Cai
et al., 2020). This realization has spurred further ex-
perimentation in the field. Interestingly, theoretical
work by L. Valiant in the 1980s (Valiant, 1984), con-
firmed by Schapire in the 1990s (Schapire, 1990),
and subsequently popularized by Surowiecki in 2004
(Surowiecki, 2004), suggests that multiple weak clas-
sifiers may perform as well as or even better than a
single strong classifier. Building on this idea, Neural
Network Ensembles (NNE) have emerged as a pow-
erful ML model design. NNEs involve ensemble clas-
368
Aguilar–Ruiz, J. and Fratini, M.
Random Neural Network Ensemble for Very High Dimensional Datasets.
DOI: 10.5220/0012763800003756
Paper published under CC license (CC BY-NC-ND 4.0)
In Proceedings of the 13th International Conference on Data Science, Technology and Applications (DATA 2024), pages 368-375
ISBN: 978-989-758-707-8; ISSN: 2184-285X
Proceedings Copyright © 2024 by SCITEPRESS – Science and Technology Publications, Lda.

sifiers that combine predictions from neural networks
trained on the original dataset, which offers another
promising approach to classification problems. How-
ever, when the number of variables is extremely large,
training many networks is prohibitively time consum-
ing. Thus, reducing the size of each of the neural net-
work units that comprise the ensemble could be a vi-
able alternative for an effective classification model.
Several ML algorithmic solutions are now avail-
able for gene expression classification problems.
However, for the sake of brevity, this work focus on
two main components of the approach presented here:
Neural Networks (NN) (Lancashire et al., 2009) and
Random Forests (RF) (Breiman, 2001). Both NN and
RF have found applications across various fields over
the years. NNs are represented by a series of layers
of neurons, whose activations depend on an activa-
tion function and connected weights, extending to the
deepest output neurons. Various architectures have
been developed for NN, such as convolutional, feed-
forward, or recurrent NN. However, a drawback is
that the number of hidden neurons and layers need
to be set beforehand, and there is usually no indica-
tion of proper configurations unless experiments have
been conducted for specific cases. Random Forests,
on the other hand, are an ensemble learning method
that constructs a multitude of decision trees (Kings-
ford and Salzberg, 2008) during training. They output
the class that is the mode of the classes (classification)
or mean prediction (regression) of the individual de-
cision trees.
The aim of this paper is to describe and exper-
imentally demonstrate that NNE can effectively re-
place existing methods in classifying very high di-
mensional biological data. Despite originating from a
theoretical concept proposed in the 1980s, this study
offers a novel focus on addressing the challenge of
very high dimensionality with an ensemble of NNs.
The model will need to address datasets with several
thousands of variables, requiring appropriate NNs ca-
pable of handling millions of parameters. However,
decomposing the problem into pieces, each of which
using lower dimensionality, could reduce the com-
plexity while maintaining the classification perfor-
mance. To achieve this goal, the principles of RF will
be employed, where several NNs will be trained and
combined in an ensemble.
Through an exhaustive analysis of the model con-
figuration, this approach also aims to outperform ex-
isting methods in the scientific literature for predict-
ing real biomedical datasets, particularly gene expres-
sion data on cancer. An experimental analysis of
the predictive power of NNE with five public cancer
datasets will be shown.
The paper will be organized as follows. Next, re-
lated approaches to the developed method will be de-
scribed. Subsequently, a more in–depth explanation
of the method will be given. Then, after the datasets
employed in the study and the design, with more
stress on the Keras architecture, will be outlined, the
results will be displayed and commented. As a sum-
mary of what has been concretely accomplished with
this study, it is safe to admit that the results achieved
for the proposed method can be reckoned to be gener-
ally positive. This statement can be made considering
the datasets on which it has been tested were classi-
fied on average more poorly by other well–known ML
models.
2 RELATED APPROACHES
The ML approaches described in this paper are in-
trinsic to the method developed and described here.
Through deep learning, these models can extract
higher–level features from raw input data. This ca-
pability provides a powerful approach, particularly in
situations where manual filtering beforehand is chal-
lenging. Deep architectures encompass various vari-
ants of fundamental approaches (Nielsen, 2018). The
diversity within these architectures has led to success
in specific domains. Deep Neural Networks (DNNs)
have demonstrated high versatility in the ML world.
Significant experimentation is dedicated to this archi-
tecture, often coupled with other robust models to im-
prove performance according to the desired solution.
Convolutional Neural Networks (CNNs) are charac-
terized by layers that perform convolutions. Typi-
cally, these layers include multiplication or other dot
product operations, pooling layers, fully connected
layers, and normalization layers. This sophisticated
architecture is particularly effective in tissue image
processing (Browne and Ghidary, 2003). Recurrent
Neural Networks (RNNs) have found applications in
analyzing biological sequences. Furthermore, other
ensembles incorporating neural networks into their
structure have been developed, with some of them
achieving high notoriety (Zhou et al., 2002). In this
work, we will focus on deep fully–connected NN.
RFs are another robust machine learning method,
suitable for both classification and regression tasks,
which has gained considerable success over the years
and paved the way for other ensemble learning meth-
ods. RF has consistently proven to be applicable and
reliable in many instances, offering reliability through
a combination of classifiers that are varied (high vari-
ance) but predict with very similar accuracy. Ensem-
bles can be developed by varying individual com-
Random Neural Network Ensemble for Very High Dimensional Datasets
369
ponents of the method, such as training data, mod-
els, and combinations. Generally, bagging, boost-
ing, and stacking are the most common techniques for
creating an ensemble (Graczyk et al., 2010). Bag-
ging (Breiman, 1996), as seen in Random Forests,
utilizes bootstrap sampling to obtain subsets of data
for training the base learners. For aggregating the
outputs of these base learners, bagging employs vot-
ing for classification tasks and averaging for regres-
sion. Boosting (Freund, 1990), exemplified by Ad-
aboost (Freund and Schapire, 1995), fits a sequence
of weak learners—models whose accuracy is slightly
better than random guessing—to weighted versions of
the data. Examples that were misclassified by ear-
lier rounds gain more weight. Predictions are then
combined through a weighted majority vote for clas-
sification or a weighted sum for regression to pro-
duce the final prediction. Stacking (Wolpert, 1992)
combines multiple classification or regression mod-
els, making it more heterogeneous than other meth-
ods, via a meta-classifier or a meta-regressor, respec-
tively. The base models are trained on a complete
training set, and then the meta-model is trained on the
outputs of the base models as features. While bag-
ging, boosting, and stacking are common techniques,
many other variants exist and can be designed to bet-
ter adapt to specific problems (Zhou, 2012).
The seminal work on ensemble of neural networks
was focused on providing a number of neural net-
works, all of them dealing with the original input size
(number of variables), in order to reduce the global er-
ror (Hansen and Salamon, 1990). It was demonstrated
that if each network can produce the right prediction
more than half the time, then the likelihood of an er-
ror by a majority decision decreased, and therefore the
ensemble error rate tended to zero when the number
of neural networks tended to infinite. This idea is in-
teresting, but replicating the original input size many
times is very time consuming, and practically unaf-
fordable in cases of very high dimensionality, such as
genomic datasets.
The subject of this study is NNE, a variant of
ensemble techniques that combines DNN architec-
tures with concepts from RF. The choice of using
NN as base classifiers in the ensemble was due the
good performance in varied domains. The strategy
employed here follows the principles of RF dataset
splitting criteria, with NN incorporation and param-
eter value selection occurring subsequently using the
same criterion as RF, as described in more detail in the
method section. In terms of computational complex-
ity, training a single large NN on high–dimensional
datasets, as included in this study, would be pro-
hibitively time–consuming due to the large number
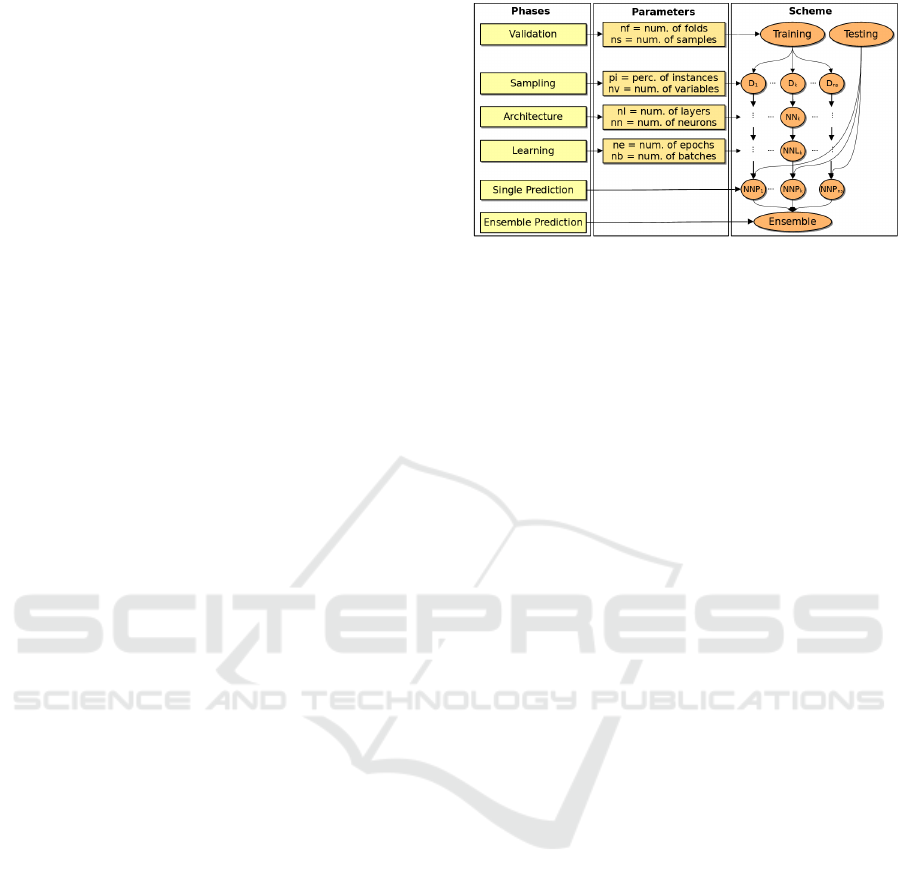
Figure 1: Neural Network Ensemble method representation.
of hyper–parameters involved. In contrast, NNE sig-
nificantly reduces cost by using smaller NNs, which
require less training time. Moreover, increasing the
number of NNs in the ensemble only incurs linear
complexity. The developed method would be advan-
tageous in contexts where other ensemble techniques
are typically applied.
3 METHOD
The principle behind NNE involves training multiple
small NNs on random subsets of the original dataset.
Even a small subset of the entire variable set can pro-
vide valuable information, and each vote in the en-
semble is decisive on its own. This approach helps
reduce bias because each perspective offers a unique
viewpoint. When dealing with very high dimensional
datasets, it is important to verify that small, precise
models can substantially contribute to the overall clas-
sification performance. This assumption is rooted in
the theory of weak learnability, as proposed by Valiant
(Valiant, 1984), which provides substantial evidence
that strong and weak learnability are equivalent con-
cepts. In essence, it suggests that a model of learnabil-
ity in which the learner needs to perform only slightly
better than random guessing is as effective as a model
in which the learner’s error can be minimized arbi-
trarily. Building upon this assumption, it is reason-
able to hypothesize that an ensemble classifier com-
posed of small weak classifiers (such as NNs) could
achieve performance equal to or better than that of a
single larger classifier (a single, huge NN), with sig-
nificantly reduced computational complexity (Shalev-
Shwartz and Singer, 2008).
The method is illustrated in Figure 1 and consists
of six phases, each involving multiple parameter set-
tings. Optimizing each parameter is crucial for max-
imizing the method’s effectiveness, but determining
the best combination within the parameter space can
be challenging. Parameter tuning is computationally
expensive and often requires testing numerous com-
DATA 2024 - 13th International Conference on Data Science, Technology and Applications
370

binations. However, optimizing the parameters of a
set of small NNs is typically less expensive than opti-
mizing a single, large NN.
Let M represent the number of variables and N
represent the number of instances in the dataset. The
parameters defining the overall structure of the model
are listed in order of implementation by the method:
• n f : number of folds.
• ns: number of samples.
• nv: number of variables at each sample.
• pi: percentage of instances.
• nl: number of layers.
• nn: number of nodes.
• ne: number of epochs.
• nb: number of batches.
The parameters n f and ns are set at the beginning for
validation. n f can be chosen according to the needs
and goals of the experiment, whereas ns requires an
automatic adjustment based on dataset dimensional-
ity evaluation. More specifically, the adjustment is
performed by means of a mathematical expression re-
lated to nv. Considering that ns would depend on
the probability of not choosing a single variable after
ns extractions with replacement, in order to guaran-
tee that the probability P of not using any variable in
the model would be minimal after ns extractions of nv
variables, the following Eq. 1, that shows the relation
between P and nv, ns and M need to be analyzed.
P =
1 −
M−1
nv−1
M
nv
!
ns
(1)
Eq. 1 can be simplified in order to relate the prob-
ability with the number of samples and the number
of variables at each sample, thus neglecting the bino-
mial coefficients, as shown in Eq. 2. As the number
of selected variables and the number of samples in-
crease, the probability tends to zero. Indeed, we found
a reasonable compromise by setting the value for P as
P =
1
√
M
(for instance, for a dataset with 30,000 genes,
P ≈ 0.005). This value represents a very low proba-
bility when the number of variables M is large, as is
often the case with genomic data.
P =
1 −
nv
M
ns
(2)
The parameter nv is defined for the sampling pha-
se, where dimensionality reduction occurs. Although
set at the beginning along with ns, it is important
to note that the splitting is executed after specifying
the value of ns. The value of nv is established by
the expression found as the splitting criterion in RF,
i.e., nv =
√
M, which has already proven to be effec-
tive with decision trees. Thus, the number of samples
could be calculated as shown in Eq. 3.
ns =
log
1
√
M
log
1 −
1
√
M
(3)
For example, the Prostate GSE6919 U95B dataset,
with 12,621 variables, would require 526 samples of
112 variables to satisfy the condition. Similarly, the
Bladder GSE31189 dataset, with 54,676 variables,
would need 1,274 samples of 234 variables.
The parameter pi plays an important role when
the number of instances in the dataset is high, such
that reducing this dimension contributes to improve
the computational cost. However, in genomic datasets
it is not common to find a large number of instances,
unlike in clinical datasets. Therefore, the parameter
pi was set to 100% for all the experimental analysis,
as the number of instances ranges from 20 to 124.
During the architecture phase, Keras was em-
ployed for designing the architecture and defining pa-
rameter values (nl, nn). The architecture was inten-
tionally kept simple by fixing the nl parameter at a
value of 2, indicating two dense layers plus an output
layer. The value of nn was derived from an expres-
sion that uses the number of input neurons calculated
in nv, as shown in Eq. 4.
nn =
√
nv
log
√
nv
(4)
The parameters nb and ne were specified for the
learning phase, which is common in deep learning
where multiple passes over the same training set may
be necessary to enhance the overall predictive power
of the model. In this case, ne was limited to 500. This
decision was based on ensuring a sufficient number of
passes over the training dataset. Although the upper
bound of 500 epochs was chosen, it was rarely nec-
essary to reach that limit due to the inclusion of an
early stopping criterion. Specifically, the minimum
delta was set to 0.005 over the training loss, with a
patience of 20 epochs (i.e., if after 20 epochs the per-
formance does not improve, then the learning stops).
With this condition, the majority of training processes
(over 95%) required between 85 and 95 epochs. The
value of nb was established to be about one sixth of
the averaged number of instances (i.e., 15).
Once a classifier was trained according to the pre-
viously explained scheme, it predicted on new ex-
amples and the respective assignments were stored.
Due to the intrinsic randomness in the method, the
same examples would likely be predicted by multiple
classifiers. Subsequently, when all predicted labels
Random Neural Network Ensemble for Very High Dimensional Datasets
371

for each example were collected, the class mode was
computed. Thus, the most frequent vote per example
was selected as the class with the highest probability
among all classifiers’ votes.
The advantages of the NNE over any deep fully–
connected NN is notorious regarding the ability of
learning in local subspaces, which is much less ex-
pensive.
4 EXPERIMENTAL ANALYSIS
4.1 Datasets
The study includes five datasets on gene expression
profiles data from various types of cancers in hu-
man patients. In these datasets, genes represent the
variables, with cells containing the expression values,
while samples represent observations of sets of vari-
ables. The raw data underlying the datasets was ob-
tained using microarray technology and deposited in
the Gene Expression Omnibus (GEO) repository by
other authors. Prior to the processing phase described
in this paper, the collected datasets underwent prepro-
cessing by manual curation. This preprocessing in-
volved considerations such as sample quality assess-
ment, removal of unwanted probes, background cor-
rection, and normalization. All relevant information
regarding to the datasets is summarized in Tab. 1,
where names denote the anatomical section where the
cancer is localized, with appended codes representing
their indexing in the GEO database. For further de-
tails about the datasets, interested readers can refer to
the Structural Bioinformatics and Computational Bi-
ology Lab (SBCB Lab) website (Feltes et al., 2019).
The Colorectal GSE44861 dataset presents 105
samples, 22,278 genes and 2 classes,53 normal and 47
tumoral state classes. The Breast GSE59246 dataset
has 101 samples, 36,623 genes and 2 classes iden-
tifying a DCIS (Ductal Carcinoma In Situ) and a
IBC (Invasive Breast Cancer) respectively, two types
of cancer interesting the breast section. 45 patients
were diagnosed with DCIS and 56 with IBC. The
Bladder GSE31189 dataset has 85 samples, 54,676
genes, a tumoral urothelial and a normal urothelial
class. 34 normal against 51 tumoral patients could be
collected overall, showing the dataset certain imbal-
ance. The Renal GSE53757 dataset has only 20 sam-
ples, 22,284 genes and CCRCC (clear cell renal cell
carcinoma) class along with the normal state class.
It represents the most balanced dataset with 10 pa-
tients in normal and tumoral states, respectively. The
Prostate GSE6919 U95B dataset includes 124 sam-
ples, 12,621 genes and primary prostate tumor class
representing the disease condition and a normal state
class. Higher class balance can be achieved in this
case, with 60 patients in normal conditions and 64
suffering from prostate cancer. In general, it is quite
difficult to learn in high–dimensional spaces from so
few instances without overfitting. The diversity pro-
vided by the NNE model helps to mitigate this issue.
4.2 Design
The method integrated a NN architecture as training
components (base classifiers) and compared them to
accelerate computational time while improving good
performance. In order to make fair comparisons be-
tween NNE and a single NN, many parameters were
set the same manner. However, the NNE architec-
ture, as described in previous section, is composed of
a number of very simple NN units, instead of a unique
large NN architecture.
Multi–Layer Perceptron (MLP), being the most
basic type of NN, is a reliable ML model for vari-
ous classification problems due to its extensive use in
research projects and benchmarks. In MLP training,
the RMSProp (Root Mean Square Propagation) algo-
rithm –a variant of the RProp (Resilient Propagation)
algorithm (Riedmiller and Braun, 1993)– is employed
to decrease training time.
The Keras architecture for MLP comprises two
dense layers, each with the number of nodes obtained
by Eq. 4, and employs Rectified Linear Units (ReLU)
as the activation function for the first two layers and
softmax for the output layer. ReLU activation func-
tions are commonly used in neural networks due to
their ability to speed up training by simplifying gradi-
ent computation. The softmax function, chosen for
the last layer, converts a real vector into a vector
of categorical probabilities, enabling interpretation of
results as a probability distribution.
To improve generalization, specific regularizers
were incorporated to both MLP and NNE to apply
penalties on layer parameters and activity during opti-
mization. An Elastic Net regularization, which com-
bines the features of both L1 and L2 regularization,
was selected. This regularization eliminates the limi-
tations found in L1 regularization by estimating both
the median and mean of the data to avoid overfitting.
It is considered more appropriate when the dimen-
sional data exceeds the number of samples used, mak-
ing it well–suited for the case of genomic datasets,
with few samples and many variables present.
Additionally, the Glorot normal method was cho-
sen as the initializer. NNs are known to be sensitive
to initial weight values, necessitating consideration of
more complex initializers for achieving better results.
DATA 2024 - 13th International Conference on Data Science, Technology and Applications
372
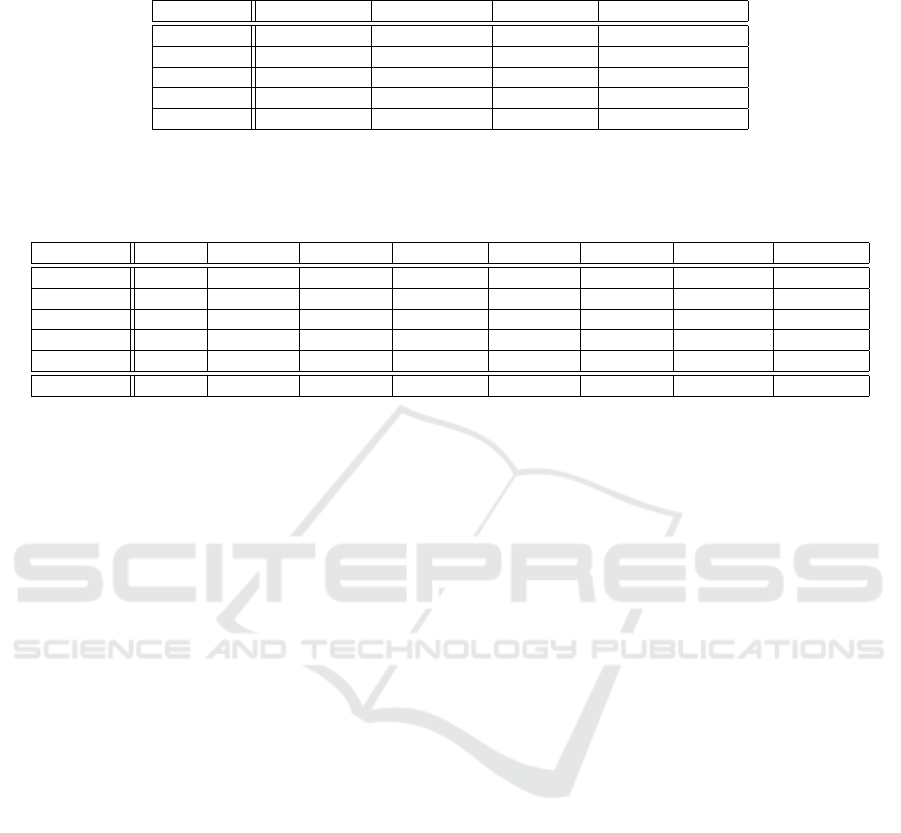
Table 1: Description of the five gene expression profile datasets on human cancer. The datasets were selected from the SBCB
Lab archive. The name, number of genes, instances, classes, and class distribution of each dataset are in columns.
Dataset No. of genes No. instances No. classes Class distribution
Colorectal 22278 105 2 53:47
Breast 36623 101 2 45:56
Bladder 54676 85 2 34:51
Renal 22884 20 2 10:10
Prostate 12621 124 2 60:64
Table 2: Description of the Neural Network–based architectures, both MLP and NNE. Var: number of variables; MLP inp:
number of input neurons for MLP; MLP hid: number of neurons in the hidden layers; MLP par: total number of parameters
for MLP; NNE inp: number of input neurons for each simple NN; NNE hid: number of neurons in the hidden layer for NNE;
NNE units: number of simple NN for the NNE; NNE par: total number of parameter for the NNE architecture.
Dataset Var. MLP inp. MLP hid. MLP par. NNE inp. NNE hid. NNE units NNE par.
Colorectal 22,278 22,278 69 1,539,127 149 11 743 110,719
Breast 36,623 36,623 84 3,085,651 191 12 1001 191,.106
Bladder 54,676 54,676 99 5,416,637 234 13 1274 298,073
Renal 22,884 22,884 55 697,579 112 10 526 58,924
Prostate 12,621 12,621 69 1,597,909 151 11 755 114,020
Mean 31,550 31,550 77 2,684,749 172 12 886 2,025,484
No constraints were specified in any layer, allowing
flexibility in model optimization.
To prevent overfitting in the MLP, the early stop-
ping technique was implemented as a form of regular-
ization, with a patience of 20 epochs (the number of
epochs with no improvement after which training will
be stopped) and a minimum delta of 0.005 (the min-
imum change in the monitored quantity to qualify as
an improvement). These values were chosen to make
fair comparisons to the NNE approach.
Given the number of input neurons for the MLP,
which incorporates quite complexity in the learning
phase, the number of epochs was set to 5,000, al-
lowing more potential room for improvement, and the
batch size was set to 15, with the training data shuffled
before each epoch.
The training sets underwent min–max normaliza-
tion, scaling the values between 0 and 1. The normal-
ization parameters obtained from the training set were
then used to normalize the test set in the same manner.
For all experiments, 3–fold cross–validation was
employed for validation, with stratified sampling used
to ensure homogeneous splitting of the classes at ev-
ery iteration. This number of folds was chosen to al-
low a fair comparison with the results published in
(Feltes et al., 2019) using the same datasets with other
classifiers. In order to compare the size of both ar-
chitectures, in terms of difficulty of parameter opti-
mization, Tab. 2 describes the complexity of both
MLP and NNE. The NNE architecture compared to
the MLP needs, on average, less amount of parame-
ters to be optimized.
Benchmarking was conducted by considering sev-
eral ML models, including Na
¨
ıve–Bayes, Random
Forest, k–Nearest Neighbors, and MLP. These models
had previously been run by other authors on the five
selected datasets, with dimensionality reduction tech-
niques such as t–SNE and PCA applied to the original
data. Evaluation of results was based on accuracy and
the area under the ROC curve as metrics.
Technically, the approach was developed on the
KNIME (Berthold et al., 2007) data analytics plat-
form, which offers a variety of functional nodes for
machine learning. Additionally, the integration of the
Keras (Chollet et al., 2015) library for deep learning
provided the necessary tools for implementing NNs
within the KNIME framework.
4.3 Results
To ease replicability, several well–known classifiers
have been chosen: Na
¨
ıve–Bayes (NB), Multi–Layer
Perceptron (MLP,) Random Forests (RF) and k–
Nearest Neighbors (kNN). All these ML algorithms
are commonly employed in microarray gene expres-
sion analysis, and generally present a good overall
performance. Upon running NNE on the five speci-
fied datasets with a 3–fold cross–validation, the fol-
lowing findings emerged.
For the Bladder dataset, an overall accuracy of
0.553 was recorded. The area under the ROC curve
was computed at 0.601. Notably, the accuracy for
Bladder was comparable to RF and lower than any
other considered algorithm, apart from NB, even
though the average performance did not exceed 0.64.
For to the Prostate dataset, similarly low values
were observed. The accuracy calculation yielded a
value of 0.67, in line with other methods. However,
Random Neural Network Ensemble for Very High Dimensional Datasets
373
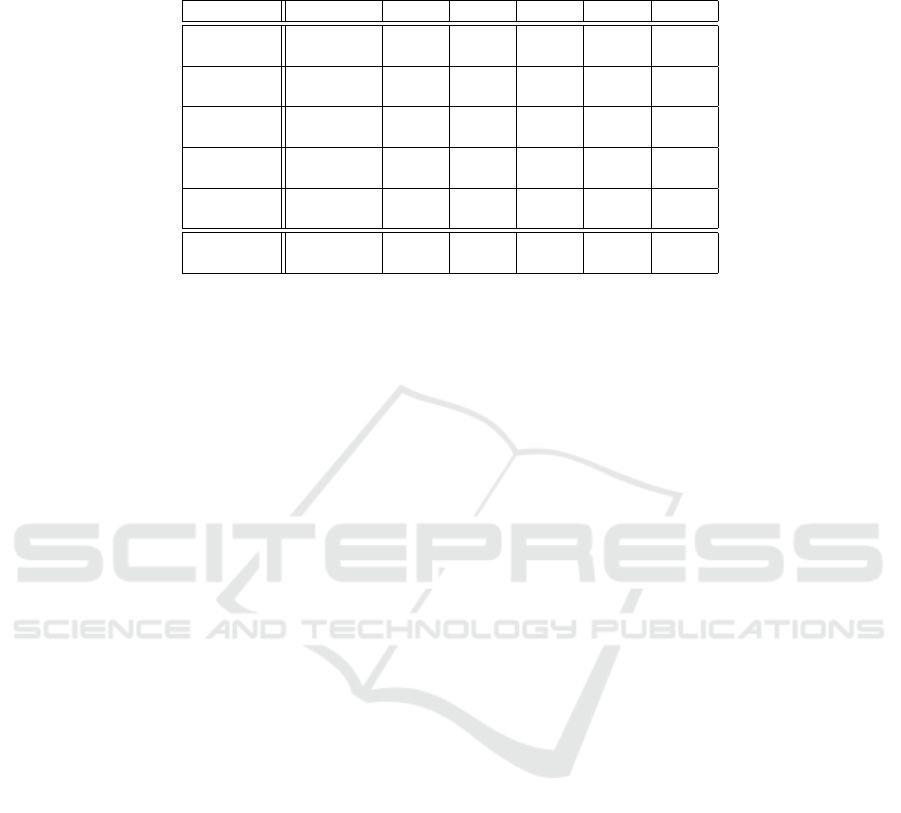
Table 3: Classification performance comparative analysis. Acc: classification accuracy; AU(ROC): area under the ROC
curve; Mean: Last row containing the Acc and AU(ROC) mean for each model over all the datasets; NNE: Neural Network
Ensemble; MLP: Multilayer Perceptron; NB: Na
¨
ıve–Bayes; RF: Random Forests; kNN: k–Nearest Neighbors.
Dataset Measure NNE MLP NB RF kNN
Colorectal
Acc
AU(ROC)
0.87
0.90
0.64
0.72
0.84
0.84
0.82
0.87
0.69
0.68
Breast
Acc
AU(ROC)
0.86
0.89
0.60
0.64
0.72
0.70
0.79
0.85
0.73
0.74
Bladder
Acc
AU(ROC)
0.55
0.60
0.58
0.50
0.46
0.46
0.55
0.53
0.62
0.63
Renal
Acc
AU(ROC)
0.85
0.85
0.80
0.86
0.90
0.90
0.85
0.89
0.85
0.80
Prostate
Acc
AU(ROC)
0.67
0.74
0.62
0.63
0.69
0.70
0.67
0.76
0.56
0.59
Mean
Acc
AU(ROC)
0.76
0.80
0.65
0.67
0.72
0.72
0.74
0.78
0.69
0.69
the area under the ROC curve reported a relatively
good value of 0.743.
In contrast, the Breast dataset displayed consider-
able improvement in metric magnitude average. Ac-
curacy was notably high at 0.861, and an area under
the ROC curve of 0.894, indicating its reliability as
a predictor. Other methods performed inferiorly to
NNE on this dataset, with accuracies below 0.8.
Likewise, the Colorectal dataset yielded results
similar to those of the Breast dataset. Accuracy was
0.867 and area under the ROC curve was 0.901. Other
methods failed to surpass NNE performance on this
dataset, remaining equal or below 0.84.
Training on the Renal dataset, despite its smaller
number of observations, led to satisfactory results.
Accuracy was recorded at 0.85, with the area under
the ROC curve reaching 0.85. Although NB outper-
formed NNE with accuracy values of 0.9, the rest re-
mained close to NNE results.
In comparison with the other algorithms selected
for benchmarking, the proposed method showed an
overall higher performance. In general, according to
related scientific literature, the choice of the classi-
fication method for such large number of genes and
small number of cases substantially influence classi-
fication success (Pirooznia et al., 2008). That means
that considering the dimensionality reduction carried
out automatically by NNE for each individual model,
the quality of these very small models, and the combi-
nation strategy, the NNE approach is suitable for very
high–dimensional contexts.
5 CONCLUSIONS
A machine learning method has been introduced for
solving classification problems, which combines the
principles of ensemble learners and neural networks
with the sampling policy of random forests. While the
concept of weak learnability equalling strong learn-
ability has been discussed extensively in the past,
placing this method in historical context, the main
features of NNE still distinguish it within the ensem-
ble landscape. Specifically, NNE addresses specific
challenges related to very high dimensionality (curse
of dimensionality) and model aggregation (overfit-
ting).
Although it is known that a single neural network
could address the problem, the very high number of
genes would result in a huge input layer, which would
make the optimization of the underlying parameters
of the architecture complex. In this work, instead, we
show that using many very small NNs, with only two
hidden layers containing few neurons, greatly reduces
the task of learning the weights, while maintaining the
quality of the classifier model.
The results obtained demonstrate that NNE can ef-
fectively classify data across the five datasets, consis-
tent with other benchmark methods (MLP, SVM, NB,
RF, and kNN). This suggests that NNE could poten-
tially replace other existing methods for classification
in the context of very high dimensional gene expres-
sion datasets. In addition, the NNE compared to the
MLP needs, on average, less amount of parameters to
be optimized.
An interesting aspect of the NNE model is that
there is scope for potential improvement. An analy-
sis of the ensemble configuration could provide even
better results by addressing two aspects: a) selecting
a variable number of input neurons for each single
NN; b) using a heuristic (rather than random) to se-
lect which input variables the NNs will use, so that
not all variables are chosen with approximately the
same probability.
DATA 2024 - 13th International Conference on Data Science, Technology and Applications
374

ACKNOWLEDGEMENTS
This work was supported by MCIN/AEI/10.13039/
501100011033 under Grant PID2020-117759GB-I00.
REFERENCES
Bellman, R. (1961). Adaptive Control Processes: A Guided
Tour. Princeton University Press.
Berthold, M. R., Cebron, N., Dill, F., Gabriel, T. R.,
K
¨
otter, T., Meinl, T., Ohl, P., Sieb, C., Thiel, K.,
and Wiswedel, B. (2007). KNIME: The Konstanz In-
formation Miner. In Studies in Classification, Data
Analysis, and Knowledge Organization (GfKL 2007).
Springer.
Breiman, L. (1996). Bagging predictors. Machine Learn-
ing, 24(2):123 – 140. Cited by: 1993; All Open Ac-
cess, Bronze Open Access.
Breiman, L. (2001). Random forests. Machine Learning,
45(1):5 – 32.
Browne, M. and Ghidary, S. S. (2003). Convolutional neu-
ral networks for image processing: An application in
robot vision. In Gedeon, T. T. D. and Fung, L. C. C.,
editors, AI 2003: Advances in Artificial Intelligence,
pages 641–652, Berlin, Heidelberg. Springer Berlin
Heidelberg.
Cai, Y., Zhang, W., Zhang, R., Cui, X., and Fang, J. (2020).
Combined use of three machine learning modeling
methods to develop a ten-gene signature for the di-
agnosis of ventilator-associated pneumonia. Medical
Science Monitor, 26.
Chollet, F. et al. (2015). Keras.
Deng, J.-L., Xu, Y.-h., and Wang, G. (2019). Identification
of potential crucial genes and key pathways in breast
cancer using bioinformatic analysis. Frontiers in Ge-
netics, 10:695.
Feltes, B. C., Chandelier, E. B., Grisci, B. I., and Dorn,
M. (2019). Cumida: An extensively curated microar-
ray database for benchmarking and testing of ma-
chine learning approaches in cancer research. Jour-
nal of Computational Biology, 26(4):376–386. PMID:
30789283.
Freund, Y. (1990). Boosting a weak learning algorithm by
majority. page 202 – 216.
Freund, Y. and Schapire, R. E. (1995). A desicion-theoretic
generalization of on-line learning and an application
to boosting. In Vit
´
anyi, P., editor, Computational
Learning Theory, pages 23–37, Berlin, Heidelberg.
Springer Berlin Heidelberg.
Graczyk, M., Lasota, T., Trawi
´
nski, B., and Trawi
´
nski, K.
(2010). Comparison of bagging, boosting and stack-
ing ensembles applied to real estate appraisal. In
Nguyen, N. T., Le, M. T., and
´
Swiatek, J., editors,
Intelligent Information and Database Systems, pages
340–350, Berlin, Heidelberg. Springer Berlin Heidel-
berg.
Hansen, L. and Salamon, P. (1990). Neural network en-
sembles. IEEE Transactions on Pattern Analysis and
Machine Intelligence, 12(10):993–1001.
Hwang, K.-B., Cho, D.-Y., Park, S.-W., Kim, S.-D., and
Zhang, B.-T. (2002). Applying Machine Learning
Techniques to Analysis of Gene Expression Data:
Cancer Diagnosis, pages 167–182. Springer US,
Boston, MA.
Kingsford, C. and Salzberg, S. (2008). What are decision
trees? Biotechnology, 26(9):1011–1012.
Lancashire, L. J., Lemetre, C., and Ball, G. R. (2009). An
introduction to artificial neural networks in bioinfor-
matics—application to complex microarray and mass
spectrometry datasets in cancer studies. Briefings in
Bioinformatics, 10(3):315–329.
Nielsen, M. A. (2018). Neural networks and deep learning.
Pirooznia, M., Yang, J., Yang, M., and Deng, Y. (2008). A
comparative study of different machine learning meth-
ods on microarray gene expression data. BMC ge-
nomics, 9 Suppl 1:S13.
Riedmiller, M. and Braun, H. (1993). A direct adaptive
method for faster backpropagation learning: the rprop
algorithm. In IEEE International Conference on Neu-
ral Networks, pages 586–591 vol.1.
Schapire, R. (1990). The strength of weak learnability. Ma-
chine Learning, 5(2):197–227.
Shalev-Shwartz, S. and Singer, Y. (2008). On the equiv-
alence of weak learnability and linear separability:
New relaxations and efficient boosting algorithms.
volume 80, pages 311–322.
Surowiecki, J. (2004). The Wisdom of Crowds: Why the
Many Are Smarter Than the Few and How Collective
Wisdom Shapes Business, Economies, Societies and
Nations. Little, Brown and Company.
Trunk, G. V. (1979). A problem of dimensionality: A sim-
ple example. IEEE Transactions on Pattern Analysis
and Machine Intelligence, PAMI-1(3):306–307.
Valiant, L. G. (1984). A theory of the learnable. Commun.
ACM, 27(11):1134–1142.
Wellinger, R. E. and Aguilar-Ruiz, J. S. (2022). A new chal-
lenge for data analytics: transposons. BioData Min-
ing, 15(1):9.
Wolpert, D. H. (1992). Stacked generalization. Neural Net-
works, 5(2):241–259.
Zhou, Z. (2012). Ensemble Methods: Foundations and
Algorithms. CHAPMAN & HALL/CRC MACHINE
LEA. Taylor & Francis.
Zhou, Z.-H., Wu, J., and Tang, W. (2002). Ensembling neu-
ral networks: Many could be better than all. Artificial
Intelligence, 137(1):239 – 263.
Random Neural Network Ensemble for Very High Dimensional Datasets
375
