
Antibiotic Resistance Gene Identification from Metagenomic Data Using
Ensemble of Finetuned Large Language Models
Syama K
a
and J. Angel Arul Jothi
b
Department of Computer Science, Birla Institute of Technology and Science Pilani Dubai Campus, Dubai, U.A.E.
Keywords:
Antibiotic Resistance Gene, Ensemble Learning, Transformer, LLM.
Abstract:
Antibiotic resistance is a potential challenge to global health. It limits the effect of antibiotics on humans. An-
tibiotic resistant genes (ARG) are primarily associated with acquired resistance, where bacteria gain resistance
through horizontal gene transfer or mutation. Hence, the identification of ARGs is essential for the treatment
of infections and understanding the resistance mechanism. Though there are several methods for ARG identi-
fication, the majority of them are based on sequence alignment and hence fail to provide accurate results when
the ARGs diverge from those in the reference ARG databases. Additionally, a significant fraction of pro-
teins still need to be accounted for in public repositories. This work introduces a multi-task ensemble model
called ARG-LLM of multiple large language models (LLMs) for ARG identification and antibiotic category
prediction. We finetuned three pre-trained protein language LLMs, ProtBert, ProtAlbert, and Evolutionary
Scale Modelling (ESM), with the ARG prediction data. The predictions of the finetuned models are combined
using a majority vote ensembling approach to identify the ARG sequences. Then, another ProtBert model is
fine-tuned for the antibiotic category prediction task. Experiments are conducted to establish the superiority of
the proposed ARG-LLM using the PLM-ARGDB dataset. Results demonstrate that ARG-LLM outperforms
other state-of-the-art methods with the best Recall of 96.2%, F1-score of 94.4%, and MCC of 90%.
1 INTRODUCTION
Antibiotics are one of the significant discoveries of
the 20th century, saving millions of lives from infec-
tious diseases. However, their widespread use and
misuse make pathogens increasingly resistant to an-
tibiotics. The World Health Organization (WHO) has
listed antibiotic resistance among the top 10 threats to
global health. Furthermore, according to WHO, an-
tibiotic resistance directly caused 1.27 million deaths
worldwide in 2019, and if no action is taken, this
number is predicted to increase to 10 million by
2050 (Murray et al., 2022; L
´
az
´
ar and Kishony, 2019).
Additionally, antibiotic resistance is spread between
pathogens by transferring antibiotic resistant genes
(ARG) through food, water, animals, and humans.
Therefore, identifying ARG in pathogens is signifi-
cant in stopping their spread, understanding the resis-
tance mechanism, and developing the targeted treat-
ment or control measures. Global efforts such as the
Global Antimicrobial Resistance Surveillance System
and the Global Antibiotic Research and Development
a
https://orcid.org/0009-0000-6297-4407
b
https://orcid.org/0000-0002-1773-8779
Partnership have been initiated for this. The pri-
mary focus of these consortium efforts is to develop
an efficient tool for identifying antibiotic resistance
(Mendelson M, 2015). Culture-based Antibiotic Sus-
ceptibility Tests (AST) are the standard practice in
clinical microbiology that determine the effectiveness
of antibiotics against specific bacteria. However, it
takes weeks to get the results and does not apply to
the unculturable bacteria (Pham and Kim, 2012).
The emergence of high-throughput DNA sequenc-
ing techniques in metagenomics helped the develop-
ment of various tools to profile the DNA of pathogens
and increased the amount of DNA and protein se-
quences in public databases. For example, UniProt
(Consortium, 2015) is the largest collection of pro-
tein sequences available after merging it with pro-
teins translated from multiple metagenomic sequenc-
ing projects. This, in turn, encouraged researchers to
enhance the understanding of the functional diversity
of microbial communities significantly. This knowl-
edge helped identify ARGs in different pathogens
present in livestock manure, compost, wastewater
treatment plants, soil, water, and the human micro-
biome (Mao et al., 2015; Pehrsson et al., 2016). How-
102
K., S. and Jothi, J.
Antibiotic Resistance Gene Identification from Metagenomic Data Using Ensemble of Finetuned Large Language Models.
DOI: 10.5220/0012999100003838
Paper published under CC license (CC BY-NC-ND 4.0)
In Proceedings of the 16th International Joint Conference on Knowledge Discovery, Knowledge Engineering and Knowledge Management (IC3K 2024) - Volume 1: KDIR, pages 102-112
ISBN: 978-989-758-716-0; ISSN: 2184-3228
Proceedings Copyright © 2024 by SCITEPRESS – Science and Technology Publications, Lda.

ever, the main challenge faced by researchers is a no-
table portion of proteins remains unannotated.
ARG identification methods are categorized into
sequence-based alignment or assembly and machine
learning (ML)-based. For alignment-based methods
(McArthur et al., 2013), the query ARG sequence
is compared against the existing ARG sequences in
the database using alignment tools such as BLAST
(Altschul et al., 1990), DIAMOND (Buchfink et al.,
2015), and BWA (Li and Durbin, 2009). Although
these methods are widely used in ARG identifica-
tion, they also have disadvantages. For example,
the sequence-based methods may miss novel genes
that are not present in the reference genome database
(Chowdhury et al., 2019), and the accurate results are
highly dependent on the value of the critical hyper-
parameter, such as the similarity threshold (Li et al.,
2021). Alternatively, multiple ML methods have been
developed for ARG identification tasks (Gibson et al.,
2015; Arango-Argoty et al., 2018a). ML-based meth-
ods depend on the features representing the character-
istics of ARGs (Rupp
´
e et al., 2019) and learn the sta-
tistical patterns of ARGs. So, ML methods are able
to identify novel genes (Li et al., 2018). However, the
ML methods are trained using the genetic features ex-
tracted from the ARG sequences of the particular or-
ganism of interest. This limits their capacity to a more
generalized applicability. Deep learning (DL) meth-
ods are especially powerful due to their inherent capa-
bility to learn features, avoiding separate feature ex-
traction. In both ML and DL methods, researchers al-
ways try to improve and optimize classification mod-
els to achieve better accuracy. Ensemble learning is
a widely used technique to enhance classification ac-
curacy (Miah et al., 2024). It aggregates two or more
base classifiers to improve the predictive performance
of the combined classifier, and it overcomes the weak-
ness of a single weak base classifier.
Presently, to uncover the properties of the novel
ARGs, the ideas embedded in natural language pro-
cessing (NLP) are adopted into protein sequence pro-
cessing. Protein sequences are considered as sen-
tences in protein language, and then NLP techniques
are used to extract the features in the protein se-
quences. In particular, transformer-based large lan-
guage models (LLM) (Devlin et al., 2018) have
achieved state-of-the-art (SOTA) performance for
several NLP and protein language tasks (Bepler and
Berger, 2021). Few LLM-based ARG identification
models have been developed (Wu et al., 2023) for
ARG identification. These models have been widely
used as feature extractors, demonstrating significant
improvements in various tasks. However, finetuning
the pre-trained model further improves the model’s
predictive power. Finetuning involves training a pre-
trained model further on a specific task or dataset
to enhance its performance for that task. Since the
model is already pre-trained on a large dataset, fine-
tuning requires significantly less time and computa-
tional resources. Hence, an ARG prediction tool that
harnesses the power of LLM-based models is in high
demand.
In this work, a multi-task ensemble model, ARG-
LLM, is used to leverage the prediction of ARG and
then further identify what antibiotic family it is resis-
tant to. It harnesses the capabilities of three publicly
available pre-trained transformer-based LLMs such as
ProtBert (Elnaggar et al., 2021), ProtAlbert (Elnag-
gar et al., 2021), and Evolutionary Scale Modelling
(ESM) (Rao et al., 2021). In the first task, the three
LLMs are finetuned with the ARG prediction dataset.
The prediction output of each of the language models
is passed through a majority-voting ensemble method.
In the second task, the ProtBert model is finetuned
with the Antibiotic category prediction dataset, and
those sequences predicted as ARGs in the first task
are further passed through the fine-tuned model for
the prediction of antibiotic categories.
This paper is organized as follows. Section 2 re-
views previous works done in ARG prediction and
Antibiotic category prediction tasks. Details of the
dataset used in this work are explained in Section 3.
Section 4 presents the methodology. The experiments
and the evaluation metrics are provided in Section 5.
The results and discussion are presented in Section
6. Section 7 provides the conclusion and the future
work.
2 RELATED WORKS
Antibiotic resistance is a serious global threat to hu-
man health that urgently requires practical action.
Identifying antibiotic resistant genes is a crucial step
in understanding the mechanism of antibiotic resis-
tance. This section covers an overview of the related
works introduced in the ARG identification field, em-
phasizing the works done using ML and DL methods.
The traditional computational methods devel-
oped for ARG identification are all sequence-
based. Hence, they are designed to identify specific
pathogens’ ARGs. For instance, ResFinder (Klein-
heinz et al., 2014) predicts specifically plasmid-borne
ARGs and the tool developed in (Bradley et al., 2015)
is dedicated to 12 types of antimicrobials. Simi-
larly, another study (Davis et al., 2016) is limited to
identifying ARGs encoding resistance to carbapenem,
methicillin, and beta-lactam antibiotics. Most of
these tools identify the query sequence’s similarity
Antibiotic Resistance Gene Identification from Metagenomic Data Using Ensemble of Finetuned Large Language Models
103

with the sequences in the existing microbial resis-
tance databases, using a ”best hit” approach to pre-
dict whether a sequence is an ARG. These methods
require a cutoff threshold to identify the similarity be-
tween the sequences. This restricts those models from
identifying novel ARGs (McArthur and Tsang, 2017).
To overcome the disadvantages of the previous meth-
ods, many ML and DL-based methods have been in-
troduced.
The work by Arango et al. (Arango-Argoty et al.,
2018b) introduced DeepARG, a novel DL approach
for predicting ARGs from metagenomic data. It con-
tained two components: DeepARG-SS for classifying
short reads and DeepARG-LS for annotating novel
ARG genes. It used a Deep Neural Network (DNN)
architecture for predicting ARGs from metagenomic
data, and a bitscore-based dissimilarity index was
used as the feature for the DL model. The DeepARG-
SS model, trained on short sequence reads, achieved
an overall precision of 0.97 and recall of 0.91 for the
30 antibiotic categories tested.
The HMD-ARG model in (Li et al., 2021) con-
sisted of hierarchically connected three DL models
that predict ARG properties by focusing on antibiotic
resistance type, mechanism, and gene mobility. Con-
volutional Neural Network (CNN) models were used
at each level. At the first level, the sequences were
classified into ARG or not. The ARG sequences were
classified in the second level based on the resistant
antibiotic family, resistant mechanism, and gene mo-
bility information. In the final level, if the predicted
antibiotic family was beta-lactamase, the framework
further predicted the subclass of beta-lactamase. The
framework could identify ARGs without querying ex-
isting databases. The HMD-ARG model achieved
an Accuracy of 0.948, Precision of 0.939, Recall of
0.951, and F1 of 0.938.
Another work named ARG-SHINE by (Wang
et al., 2021) introduced a novel ARG prediction
framework by integrating sequence homology and
functional information with DL techniques. It used
CNN for the classification. This framework proposed
the method to combine sequence homology, func-
tional information, and DL, and the integration im-
proved antibiotic resistance prediction accuracy. The
ARG-SHINE model achieved an Accuracy of 0.8557
and an F1 of 0.8595.
A recent work named PLM-ARG proposed by
(Wu et al., 2023) introduced a novel method for ARG
identification using a pre-trained protein language
model, ESM-1b. It harnessed the power of ESM-
1b to generate embedding for protein sequences and
utilized the Extreme Gradient Boosting (XGBoost)
ML model to classify the antibiotic category. The
study provided insights into applying Artificial Intel-
ligence (AI)-powered language models for ARG iden-
tification. The PLM-ARG model achieved an Accu-
racy of 0.912, Precision of 1, Recall of 0.825, F1 of
0.904, and Mathews Correlation Coefficient (MCC)
of 0.838.
The literature review shows that the efficacy of
transformer-based NLP models is less utilized in the
ARG identification task. Researchers have identified
that finetuning the transformer-based models gives an
exceptional performance in NLP tasks (Devlin et al.,
2018). However, finetuning the transformer-based
models for ARG prediction with the ARG dataset has
yet to be explored. Hence, in this work, we finetune
the protein language models and use the finetuned
model for classification. Additionally, we utilized the
capacity of ensembling the prediction of the finetuned
models to identify ARG sequences.
3 DATASET
We collected antibiotic resistance gene sequences
from the published ARG database PLM-ARGDB
(Wu et al., 2023). It contains 57158 gene sequences,
28579 of which are labeled as ARG and 28579 of
which are labeled as non-ARGs. The sequences
which are labeled as ARG are further labeled with
their antibiotic category. The 26391 ARGs in the
28579 ARG sequences are labeled with 22 explicit
resistance categories, and 2188 ARGs are tagged
with a general category “multi-drug” or “antibiotic
without defined classification.” PLM-ARGDB is con-
structed by extracting ARG sequences from six pub-
licly available ARG databases, as 4790 from CARD
(Jia et al., 2016), 859 from ResFinder (Zankari et al.,
2012), 2044 from MEGARes (Lakin et al., 2017), 444
from AMRFinderPlus (Feldgarden et al., 2019), 9863
from ARGMiner, and 10579 from HMD-ARG-DB
(Li et al., 2021). The non-ARG sequences are taken
from the UniProt database.
4 PROPOSED METHODOLOGY
In this work, we introduce a novel multi-task en-
semble framework, ARG-LLM, which automatically
identifies the ARGs and the categories of antibi-
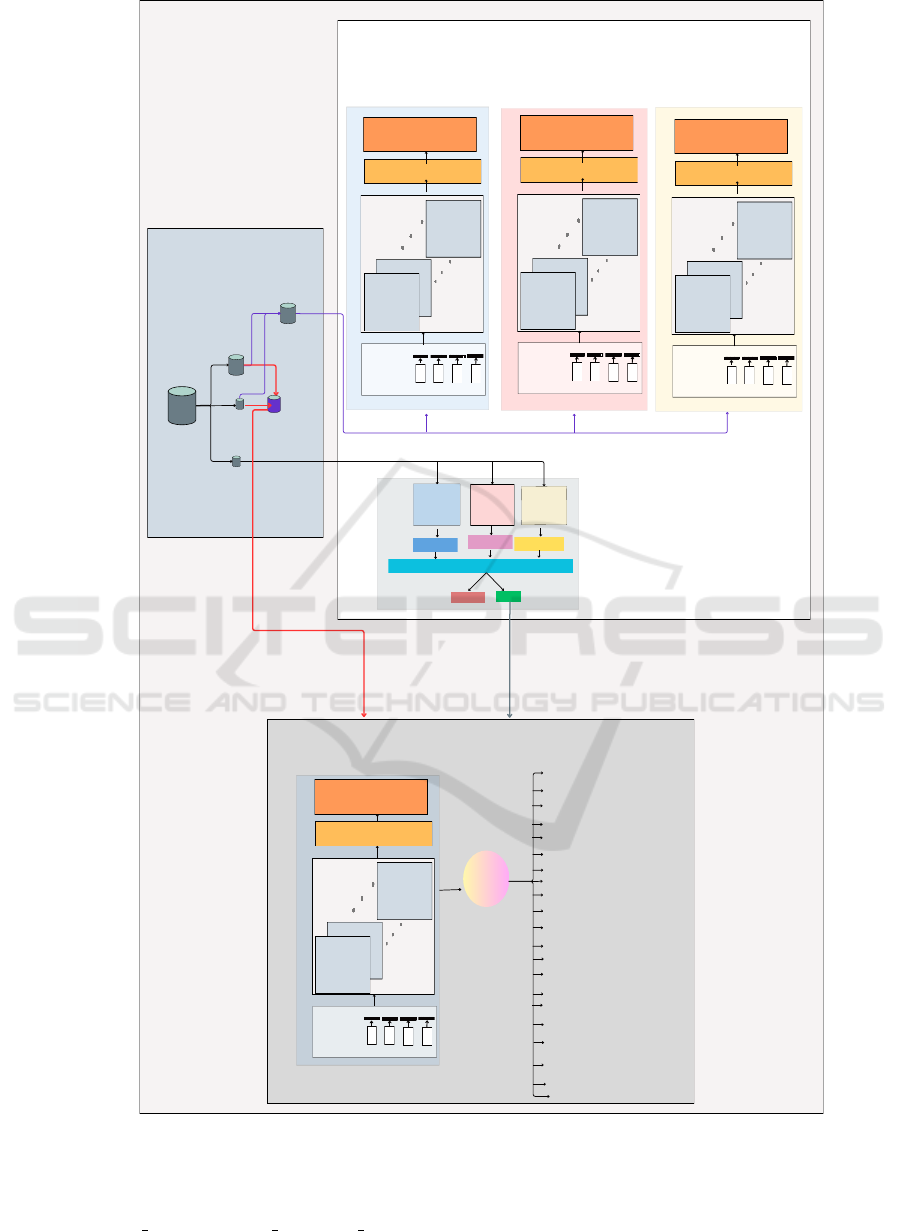
otics to which the pathogen is resistant. Figure 1
presents the overall methodology of this work. ARG-
LLM performs two tasks: one is the ARG predic-
tion task, and the other is the Antibiotic category
prediction task. ARG-LLM starts with preprocess-
ing the dataset and preparing the data for subsequent
finetuning and prediction. The ARG prediction task
KDIR 2024 - 16th International Conference on Knowledge Discovery and Information Retrieval
104
Protein
sequence
dataset
validation
set
Train set
ProtBert_bin
Test set
ProtAlbert_
bin
Prediction 1
Prediction 2
Majority voting
ESM_bin
Prediction 3
ARG
Non ARG
Ensemble Prediction
ARG prediction
dataset
categories of
antibiotic the
pathogen is
resistant to
Fine-tune the ProtBert model for
Category prediction(ProtBert_Cat)
pleuromutilin
fluoroquinolone
glycopeptide
lincosamide
peptide
phenicol
sulfonamide
sulfone
tetracycline
others
aminoglycoside
macrolide
rifamycin
beta-lactam
diaminopyrimidine
disinfecting agents and antiseptics
nitroimidazole
nucleoside
streptogramin
aminocoumarin
alkaloids with antibiotic activity
Antibiotic category Prediction task
Pretrained
ProtBert
Encoder Layer
N
Pretrained
ProtBert
Encoder Layer
1
Pre-trained Transformer model
Input sequence embedding from
last hidden layer
Classification head
S
P
G
...
Tokenization and
Encoding
Input protein
sequence
Fine-tune the
ProtAlbert model
Fine-tune the ESM model
(esm2_t12_35M_UR50D)
Fine-tune the
ProtBert model
ARG Sequences from test
data
Antibiotic category
prediction dataset
ARG Prediction task
Finetuning with Antibiotic
category prediction dataset
Preprocessing
Pretrained
ProtBert
Encoder Layer
N
Pretrained
ProtBert
Encoder Layer
1
Pre-trained Transformer model
Input sequence embedding from
last hidden layer
Classification head
S
P
G
...
Tokenization and
Encoding
Input protein
sequence
Pretrained
ProtAlbert
Encoder Layer
N
Pretrained
ProtAlbert
Encoder Layer
1
Pre-trained Transformer model
Input sequence embedding from
last hidden layer
Classification head
S
P
G
...
Tokenization and
Encoding
Input protein
sequence
Pretrained
ESM
Encoder Layer
N
Pretrained
ESM
Encoder Layer
1
Pre-trained Transformer model
Input sequence embedding from
last hidden layer
Classification head
S
P
G
...
Tokenization and
Encoding
Input protein
sequence
Figure 1: Overview of the proposed methodology.
finetunes the pre-trained base LLM models, such as
ProtBert, ProtAlbert, and ESM. Then, these finetuned
models (ProtBert bin, ProtAlbert bin, ESM bin) are
used as base classifiers for the majority voting classi-
fier to predict whether the given sequence is ARG or
not. The Antibiotic category prediction task finetunes
the ProtBert model and then predicts the categories of
antibiotics for those sequences that are predicted as
Antibiotic Resistance Gene Identification from Metagenomic Data Using Ensemble of Finetuned Large Language Models
105

ARG during the ARG prediction task. The following
subsections explain each step in detail.
4.1 Preprocessing
In this step, the sequences are read from the database
which is in the format of a FASTA file. The ARG
sequences labeled ”multi-drug” or ”antibiotic without
defined classification” are changed to the label ”oth-
ers”. Thus, the sequences have two labels, where one
is the ARG label and the other 21 are the antibiotic
categories label. The ARG label is given as 0 or 1,
where 0 represents non-ARG and 1 represents ARG.
The antibiotic category labels are present for only
those sequences with ARG equal to 1. The antibiotic
category labels are transformed into a binary matrix
format using sklearn MultiLabelBinarizer(). Then,
separate train and validation sets are formed, one for
the binary (ARG) prediction and the other for the mul-
tilabel (Antibiotic category) prediction. Hence, in this
work, we refer to the ARG prediction dataset as the
training and validation datasets used for ARG pre-
diction. These datasets contain only the protein se-
quences and their ARG labels. Furthermore, these
datasets are used to finetune the three base LLMs.
Similarly, we refer to the Antibiotic category predic-
tion dataset as the train and the validation datasets
used for Antibiotic category prediction. This dataset
contains the protein sequences and their Antibiotic
category labels, which are used to finetune the Prot-
Bert model for Antibiotic category prediction.
4.2 Architecture of ARG-LLM
The two tasks of ARG-LLM are explained in the fol-
lowing subsections.
4.2.1 ARG Prediction
ARG prediction task includes finetuning the base
LLM models with ARG prediction dataset, and
combine the predictions done by the finetuned model
using ensemble prediction.
a) Finetuning the LLMs:
This task utilized three transformer-based LLMs.
The transformer model was introduced in 2017
by Vaswani et al. (Vaswani et al., 2017). It is a
neural network model that understands the context
of the input sequence. Usually, the transformer
has an encoder-decoder architecture. However, the
pre-trained models used in this study are based
on Encoder-only Transformer (EOT) architecture
because they focus on generating embedding for the
protein sequences. EOT understands the features
S
P
G
...
Feedforward
Add&Norm
Add&Norm
Multi-Head
attention
Tokenization and
Encoding
Input protein sequence
Representation
A single layer of encoder
Query key
Value
Figure 2: Architecture of a single layer of transformer en-
coder.
and patterns in the input sequence and generates a
representation for the input. The encoder is a stack of
multiple layers. The encoder takes the input protein
sequences composed of amino acids, passes them
through a series of operations, and generates the
abstract representation that encapsulates the learned
information from the entire sequence.
Figure 2 shows a single encoder layer in the trans-
former. It comprises of three modules: tokeniza-
tion and encoding module, self-attention module, and
feed-forward module. Tokenization aims to tokenize
each amino acid (word) in the protein sequence (sen-
tence). Then, the encoding step converts each token
to a vector. In order to provide information about the
position of a token in the sequence, positional encod-
ing is then added to the vector of each token. Since
transformers lack an inherent sense of sequence or-
der, positional encoding is necessary to add informa-
tion about the order of tokens in each sequence. All
the pre-trained models used in this study use absolute
positional encoding (Vaswani et al., 2017). Absolute
positional encoding uses sine and cosine functions to
generate a unique vector for each token’s fixed posi-
tion in the sequence. These vectors are added to the
input representations of amino acids before being fed
into the transformer layers. The positional encoding
for each position pos pos is calculated as follows.
PE(pos,2i) = sin(
pos
10000
2i/d
model
)
PE(pos,2i + 1) = cos(
pos
10000
2i/d
model
)
(1)
where i is the dimension of the positional encoding,
d
m
odel is the dimensionality of the encoded input.
The self-attention module consists of self-
attention and layer normalization. Self-attention
KDIR 2024 - 16th International Conference on Knowledge Discovery and Information Retrieval
106

uses a multi-head attention mechanism to relate each
amino acid in the input protein sequence with other
amino acids. The encoded vector of each amino acid
(token) is then fed to three parameters: Query (Q),
Key (K), and Value (V). Q is a vector representing the
token for which the attention scores are calculated. K
are vectors associated with each token in the sequence
and are used to compare against the Q vector to com-
pute a score. V are vectors the same as K but are used
to calculate the final representation of the word after
the attention mechanism is applied. In a multi-head
attention mechanism with h heads, the Q, K, and V
are linearly projected, and h versions of Q, K, and V
are obtained as follows.
Q
i
= XW
Q
i
; K
i
= XW
K
i
; V
i
= XW
V
i
(2)
where i = 1,2,···,h,
W
Q
i
,W
K
i
, and W
V
i
are the learned projection matrices
for head i, and X is the input tokens matrix.
Each attention head i performs a scaled dot prod-
uct attention as follows.
Attention(Q
i
,K
i
,V
i
) = so f tmax(
Q
i
K
T
i
√
d
k
)V
i
(3)
where d
k
is the dimension of the Key vectors.
After computing the attention from all the heads,
the attention vectors are concatenated and trans-
formed using a linear transformation as given below.
MultiHead(Q, K,V) = concat(head
1
,head
2
,···,
head
h
)W
O
(4)
where head
i
= Attention(Q
i
,K
i
,V
i
), and W
O
is the
learned weight matrix for linear transformation.
By computing attention scores across multiple
heads and combining the results, the transformer
model can better understand the context and depen-
dencies within the data. The output of the multi-head
attention is added to the input using the residual con-
nection, and the sum is passed to the layer normaliza-
tion operation.
The output of the self-attention module is passed
to the feed-forward module. The feed-forward mod-
ule consists of a fully connected feed-forward net-
work containing two linear transformations with a
ReLU activation in between. Equation 5 shows the
feed-forward network operation performed on input
x.
FFN(x) = ReLU (W
1
x + b
1
)W
2
+ b
2
(5)
where W
1
, W
2
are the learned weight matrices, and b
1
,
b
2
are biases.
The output of the feed-forward module is added
to its input using a residual connection, followed by
layer normalization. These operations are performed
in each of the layers of the encoder. The transformer
encoder can have N such layers. The output of the
final encoder layer is the abstract representation of the
input sequence with a rich contextual understanding.
After the success of transformers in many NLP
tasks, Devlin et al. introduced a bidirectional Encoder
Only transformer called Bidirectional Encoder Rep-
resentations from Transformers (BERT) for text pro-
cessing in 2018 (Devlin et al., 2018). BERT differs
from traditional transformer models by using a bidi-
rectional approach, meaning it considers the context
from both the left and right sides of a sequence. BERT
is pre-trained on a large corpus of text using two unsu-
pervised tasks: Masked Language Modeling (MLM)
(Taylor, 1953) and Next Sentence Prediction (NSP).
BERT can be adapted to various NLP tasks by adding
a simple output layer. The models used in this work,
such as ProtBert, ProtAlbert, and ESM, are based on
the BERT architecture.
ProtBert: It is a protein-specific variant of BERT
1
developed by training the pre-trained BERT model
using 393 billion amino acid sequences from UniRef
(Suzek et al., 2015) and BFD(Steinegger and S
¨
oding,
2018) databases. It is trained using MLM objective
in a self-supervised manner. The number of layers
of ProtBert was increased to 30 compared to BERT,
which had 24 layers.
ProtAlbert: It is a protein-specific variant of A Lite
BERT(ALBERT
2
) model developed by pretraining
the Albert model using UniRef100 (Suzek et al.,
2015) dataset. Albert models use parameter sharing
across layers, which reduces the total number of pa-
rameters while maintaining a similar model depth,
making it a Lite version of BERT.
ESM: It is a transformer-based model designed ex-
plicitly for protein sequence analysis and was devel-
oped by Meta AI (formerly Facebook AI Research).
ESM is trained with UniRef50 (Suzek et al., 2015), a
massive dataset of 250 million protein sequences en-
compassing 86 billion amino acids. The model uti-
lizes unsupervised learning to learn representations
that capture biological properties and evolutionary di-
versity from sequence data. It comes in different vari-
ants based on the number of parameters and layers. In
this work, we used ”esm2 t12 35M UR50D”, which
refers to a specific variant or configuration of the ESM
model.
To finetune the pre-trained LLMs for the ARG
prediction task, the model is modified by adding a
1
https://github.com/google-research/bert
2
https://github.com/google-research/albert
Antibiotic Resistance Gene Identification from Metagenomic Data Using Ensemble of Finetuned Large Language Models
107

Dense layer (relu)
Droput layer
Dense layer (relu)
Droput layer
Dense layer
(Softmax)
Figure 3: Architecture of the classification head.
classification head on top of the model architecture.
Figure 3 presents the architecture of classification
head. It includes two fully connected layers with a
ReLU activation function. Each fully connected layer
is followed by a dropout layer. Finally, a softmax ac-
tivation function is used for the classification.
Then we train the entire model with the ARG
prediction dataset. During training, only the weights
of the last k layers of the pre-trained model and the
newly added classification head are updated based on
the loss calculated from the classification task. The
loss function used here is the Binary Cross Entropy
(BCE) loss. After finetuning, the finetuned models
are called ProtBert bin, ProtAlbert bin, and ESM bin
respectively.
b) ARG Prediction using Ensemble of Finetuned
LLMs:
The finetuned models are used for prediction with
the test data. The predictions furnished by the
models mentioned above are combined through a
process known as majority voting (Dietterich, 2000).
This entails tallying the occurrences of ARG and
non-ARG labels. The final prediction is obtained
depending on the votes achieved by each label. For
a given protein sequence x, base classifier h
i
, each
h
i
produces a predicted class label h
i
(x), then the
majority voting method can be performed as follows.
ˆy = argmax
c∈C
N
∑
i=1
1(h
i
(x) = c) (6)
where C = {ARG, non-ARG}; set of possible class
labels, N=3 is the number of base classifiers, 1 is the
indicator function, which return 1 if the argument is
true, argmax selects the class with the maximum vote
and ˆy is the final predicted class label.
4.2.2 Antibiotic Category Prediction
In this task, a ProtBert model with a classification
head for predicting the antibiotic categories of the
ARG sequences is finetuned with the Antibiotic cate-
gory prediction dataset. The finetuned model is called
ProtBert cat. Then, ProtBert cat is used to predict
the antibiotic categories of those sequences which are
predicted as ARG by the ensemble model.
5 EXPERIMENTAL SETUP AND
EVALUATION METRICS
5.1 Experimental Setup
The proposed framework is written in Python 3, and
the libraries used are Sklearn version 1.0.2 and Py-
torch version 1.13. All the experiments are executed
on an ml.g5.xlarge instance type in Amazon Sage-
Maker, equipped with an NVIDIA A10G Tensor Core
GPU and 24 GB dedicated memory. Table 1 presents
the parameters used by each LLM.
5.2 Evaluation Metrics
The performance of the proposed model is evaluated
using metrics like: F1-score (F1), accuracy, precision,
recall and Matthews Correlation Coefficient (MCC).
Let TP, TN, FP, and FN be the number of true pos-
itives, true negatives, false positives, and false neg-
atives, respectively, then each of the metrics is cal-
culated as follows. For the multilabel classification
of category prediction the model performance was
calculated based on micro-averages for each perfor-
mance metric. Each of the metric is calculated as
shown in equation 7.
Accuracy =
T P + T N
T P + FP + T N +FN
Recall =
T P
T P + FN
Precision =
T P
T P + FP
F1 −score =
2 ×Precision×Recall
Precision + Recall
MCC =
T P ×T N −FP ×FN
p
(T P + FP)(T P + FN)(T N + FP)(T N + FN)
(7)
6 RESULTS AND DISCUSSIONS
This section presents and discusses the results of the
proposed ensemble framework obtained on the test
dataset.
KDIR 2024 - 16th International Conference on Knowledge Discovery and Information Retrieval
108
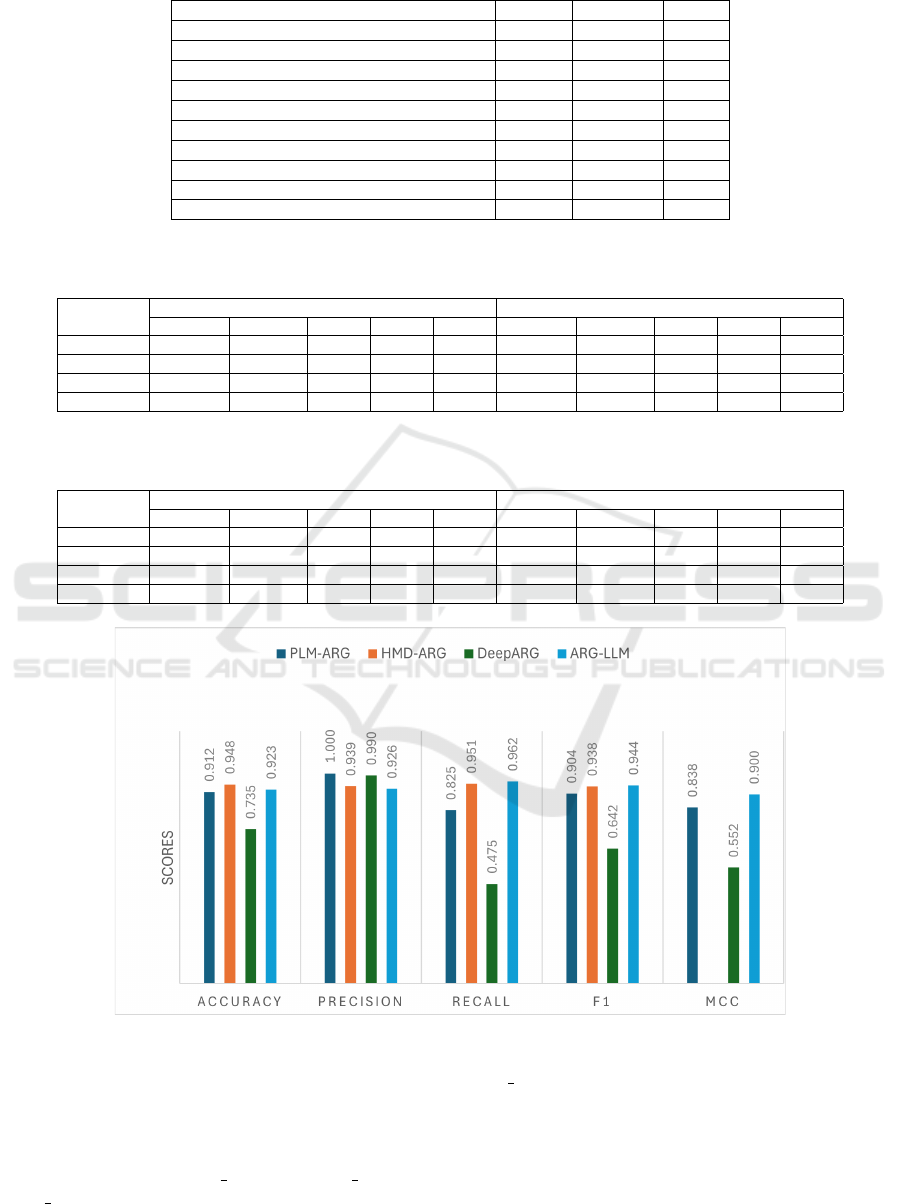
Table 1: The parameters and configurations used by each model.
Parameters ProtBert ProtAlbert ESM
Number of Layers 30 12 12
Embedding Size 1024 4096 4096
Number of Parameters 420 M 224 M 35 M
Learning rate 0.0005 0.0005 0.0005
Optimizer Adam Adam Adam
Batch size 1 1 1
1st dense layer size in the classification head 512 512 512
2nd dense layer size in the classification head 128 128 128
number of unfrozen layers 8 5 8
Loss function BCE BCE BCE
Table 2: Comparison results of individual finetuned LLMs and ARG-LLM on ARG and Antibiotic category prediction (Best
results are highlighted in bold).
ARG Prediction Category Prediction
Accuracy Precision Recall F1 MCC Accuracy Precision Recall F1 MCC
ProtBert 0.9827 0.9689 0.9753 0.9721 0.9712 0.9168 0.9324 0.9289 0.9306 0.8754
ProtAlbert 0.9752 0.9638 0.9747 0.9692 0.9624 0.9175 0.9461 0.9293 0.9376 0.8854
ESM 0.9832 0.9723 0.9859 0.9791 0.9763 0.9281 0.9085 0.9612 0.9343 0.8967
ARG-LLM 0.9931 1 0.9859 0.9929 0.9862 0.9232 0.9261 0.9616 0.9435 0.9001
Table 3: Comparison with Pre-trained LLM models as embedding generator and XGBoost as classifier for ARG and Antibiotic
category prediction (Best results are highlighted in bold).
ARG Prediction Category Prediction
Accuracy Precision Recall F1 MCC Accuracy Precision Recall F1 MCC
ProtBert 0.9562 0.9678 0.9587 0.9632 0.9215 0.9108 0.9075 0.9151 0.9113 0.8941
ProtAlbert 0.9487 0.9758 0.9475 0.9614 0.9245 0.9012 0.9161 0.9327 0.9243 0.8995
ESM 0.9923 0.9954 0.9852 0.9902 0.9758 0.9174 0.9167 0.9296 0.9231 0.789
ARG-LLM 0.9931 1 0.9859 0.9929 0.9862 0.9232 0.9261 0.9616 0.9435 0.9001
Figure 4: Comparison of the performance of ARG-LLM with state-of-the-art approaches on Antibiotic category prediction.
6.1 Comparison with Individual
Finetuned LLMs
Experiments are conducted using the individual
finetuned models ProtBert bin, ProtAlbert bin, and
ESM bin separately for ARG prediction and the Prot-
Bert cat model for category prediction. The results
achieved by each individual finetuned model are com-
pared with those of ARG-LLM. Table 2 presents
the comparison results. The results show that the
ARG-LLM invariably delivered competitive results
across multiple metrics, highlighting its ability to pre-
dict ARG and its categories. The proposed model
Antibiotic Resistance Gene Identification from Metagenomic Data Using Ensemble of Finetuned Large Language Models
109

achieved the best accuracy of 0.9931, Precision of 1,
Recall of 0.9859, F1 of 0.9929, and MCC of 0.9862
for the ARG prediction task. For the Antibiotic cat-
egory prediction task, the ESM model achieves the
best accuracy with a value of 0.9281, and the Pro-
tAlbert model achieves the best Precision of 0.9461.
ARG-LLM achieves the best Recall, F1, and MCC
with values 0.9616, 0.9435, and 0.9001, respectively.
Additionally, the ESM model’s performance is signif-
icant out of the three transformer models, as it consis-
tently shows strong prediction capability.
6.2 Comparison with Pre-Trained
LLMs as Embedding Generators
In this experiment, the pre-trained ProtBert, Pro-
tAlbert, and ESM models are used to generate em-
beddings for the sequences in the dataset. Then,
the embeddings are provided as input for a subse-
quently trained XGBoost model for ARG prediction.
A trained multilabel XGBoost classifier is used to pre-
dict the antibiotic categories. The comparison results
are presented in Table 3. From the table 3, it is evident
that the ARG-LLM achieves the best performance on
the ARG prediction task with an Accuracy of 0.9931,
Precision of 1, Recall of 0.9859, F1 of 0.9929, and
MCC of 0.9862. Similarly, ARG-LLM outperforms
the antibiotic category prediction task with best Accu-
racy of 0.9232, Precision of 0.9262, Recall of 0.9616,
F1 of 0.9435, and MCC of 0.9001.
6.3 Comparison with SOTA Methods
Figure 4 compares ARG-LLM results with SOTA
methods like DeepArg (Arango-Argoty et al., 2018b),
HMD-ARG (Li et al., 2021), and PLM-ARG (Wu
et al., 2023) for Antibiotic category prediction. The
referenced studies have not provided the results of
ARG prediction, so we are unable to give a com-
parison of ARG prediction in this section. Also, the
referenced research HMD-ARG did not present the
MCC value in their work paper; thus, we are unable
to include it in our comparison. From the available
results, it can be observed that the highest accuracy
of 0.948 is achieved by the HMD-ARG model, and
the PLM-ARG model achieves the highest precision
of 1. However, ARG-LLM achieves the best Recall,
F1, and MCC of 0.962, 0.944, and 0.900, respectively.
Additionally, ARG-LLM achieves the 2nd highest ac-
curacy of 0.923. Overall, if F1 is taken as a metric,
ARG-LLM outperforms other SOTA methods.
Overall, this work aimed to predict ARG and
antibiotic resistance categories using an ensemble
of finetuned transformer-based LLMs. The exper-
imental results reveal promising performance gains
achieved by the ARG-LLM framework. The results
from Table 3 show that finetuning the pre-trained
LLMs improves their performance in classifying the
ARG sequences into their antibiotic categories. Fine-
tuning helps the model to adapt to the specific charac-
teristics of a new, smaller dataset relevant to the target
task. Similarly, from Table 2, it is clear that ensem-
bling the three LLMs led to a significant improvement
in performance.
7 CONCLUSION
We propose a multi-task ensemble model of fine-
tuned LLMs to leverage the prediction of ARG and
then further identify what antibiotic family it is re-
sistant to. The experimental results confirm the reli-
ability of the proposed model in identifying ARGs.
The comparison results show that finetuning a pre-
trained model with a task-specific dataset improves
the model’s performance. Additionally, ensemble
prediction with the fine-tuned LLMs further enhanced
the performance of the proposed model. The out-
comes of this experimentation have powerful impli-
cations for researchers and practitioners engaged in
ARG identification tasks. The proposed model can be
a powerful tool to alleviate the global threat of antibi-
otic resistance. In the future, the ARG structural in-
formation can be incorporated with the sequence fea-
tures to improve the performance of the model.
REFERENCES
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and
Lipman, D. J. (1990). Basic local alignment search
tool. Journal of molecular biology, 215(3):403–410.
Arango-Argoty, G., Garner, E., Pruden, A., Heath, L. S.,
Vikesland, P., and Zhang, L. (2018a). Deeparg: a deep
learning approach for predicting antibiotic resistance
genes from metagenomic data. Microbiome, 6:1–15.
Arango-Argoty, G., Garner, E., Pruden, A., Heath, L. S.,
Vikesland, P., and Zhang, L. (2018b). Deeparg: a deep
learning approach for predicting antibiotic resistance
genes from metagenomic data. Microbiome, 6:1–15.
Bepler, T. and Berger, B. (2021). Learning the protein lan-
guage: Evolution, structure, and function. Cell sys-
tems, 12(6):654–669.
Bradley, P., Gordon, N. C., Walker, T. M., Dunn, L., Heys,
S., Huang, B., Earle, S., Pankhurst, L. J., Anson,
L., De Cesare, M., et al. (2015). Rapid antibiotic-
resistance predictions from genome sequence data for
staphylococcus aureus and mycobacterium tuberculo-
sis. Nature communications, 6(1):10063.
KDIR 2024 - 16th International Conference on Knowledge Discovery and Information Retrieval
110

Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and
sensitive protein alignment using diamond. Nature
methods, 12(1):59–60.
Chowdhury, A. S., Call, D. R., and Broschat, S. L. (2019).
Antimicrobial resistance prediction for gram-negative
bacteria via game theory-based feature evaluation.
Scientific reports, 9(1):14487.
Consortium, U. (2015). Uniprot: a hub for protein informa-
tion. Nucleic acids research, 43(D1):D204–D212.
Davis, J. J., Boisvert, S., Brettin, T., Kenyon, R. W., Mao,
C., Olson, R., Overbeek, R., Santerre, J., Shukla, M.,
Wattam, A. R., et al. (2016). Antimicrobial resis-
tance prediction in patric and rast. Scientific reports,
6(1):27930.
Devlin, J., Chang, M.-W., Lee, K., and Toutanova, K.
(2018). Bert: Pre-training of deep bidirectional trans-
formers for language understanding.
Dietterich, T. G. (2000). Ensemble methods in machine
learning. In International workshop on multiple clas-
sifier systems, pages 1–15. Springer.
Elnaggar, A., Heinzinger, M., Dallago, C., Rihawi, G.,
Wang, Y., Jones, L., Gibbs, T., Feher, T., Angerer, C.,
Steinegger, M., Bhowmik, D., and Rost, B. (2021).
Prottrans: Towards cracking the language of life’s
code through self-supervised deep learning and high
performance computing.
Feldgarden, M., Brover, V., Haft, D. H., Prasad, A. B.,
Slotta, D. J., Tolstoy, I., Tyson, G. H., Zhao, S., Hsu,
C.-H., McDermott, P. F., et al. (2019). Validating the
amrfinder tool and resistance gene database by using
antimicrobial resistance genotype-phenotype correla-
tions in a collection of isolates. Antimicrobial agents
and chemotherapy, 63(11):10–1128.
Gibson, M. K., Forsberg, K. J., and Dantas, G. (2015).
Improved annotation of antibiotic resistance determi-
nants reveals microbial resistomes cluster by ecology.
The ISME journal, 9(1):207–216.
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo,
P., Tsang, K. K., Lago, B. A., Dave, B. M., Pereira,
S., Sharma, A. N., et al. (2016). Card 2017: expan-
sion and model-centric curation of the comprehensive
antibiotic resistance database. Nucleic acids research,
page gkw1004.
Kleinheinz, K. A., Joensen, K. G., and Larsen, M. V.
(2014). Applying the resfinder and virulencefinder
web-services for easy identification of acquired antibi-
otic resistance and e. coli virulence genes in bacterio-
phage and prophage nucleotide sequences. Bacterio-
phage, 4(2):e27943.
Lakin, S. M., Dean, C., Noyes, N. R., Dettenwanger, A.,
Ross, A. S., Doster, E., Rovira, P., Abdo, Z., Jones,
K. L., Ruiz, J., et al. (2017). Megares: an antimicro-
bial resistance database for high throughput sequenc-
ing. Nucleic acids research, 45(D1):D574–D580.
L
´
az
´
ar, V. and Kishony, R. (2019). Transient antibiotic
resistance calls for attention. Nature microbiology,
4(10):1606–1607.
Li, H. and Durbin, R. (2009). Fast and accurate short read
alignment with burrows–wheeler transform. bioinfor-
matics, 25(14):1754–1760.
Li, Y., Wang, S., Umarov, R., Xie, B., Fan, M., Li, L., and
Gao, X. (2018). Deepre: sequence-based enzyme ec
number prediction by deep learning. Bioinformatics,
34(5):760–769.
Li, Y., Xu, Z., Han, W., Cao, H., Umarov, R., Yan, A.,
Fan, M., Chen, H., Duarte, C. M., Li, L., et al. (2021).
Hmd-arg: hierarchical multi-task deep learning for an-
notating antibiotic resistance genes. Microbiome, 9:1–
12.
Mao, D., Yu, S., Rysz, M., Luo, Y., Yang, F., Li, F., Hou,
J., Mu, Q., and Alvarez, P. (2015). Prevalence and
proliferation of antibiotic resistance genes in two mu-
nicipal wastewater treatment plants. Water research,
85:458–466.
McArthur, A. G. and Tsang, K. K. (2017). Antimicrobial
resistance surveillance in the genomic age. Annals of
the New York Academy of Sciences, 1388(1):78–91.
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad,
M. A., Baylay, A. J., Bhullar, K., Canova, M. J.,
De Pascale, G., Ejim, L., et al. (2013). The compre-
hensive antibiotic resistance database. Antimicrobial
agents and chemotherapy, 57(7):3348–3357.
Mendelson M, M. M. (2015). The world health organization
global action plan for antimicrobial resistance. S Afr
Med J, 105(5):325.
Miah, M. S. U., Kabir, M. M., Sarwar, T. B., Safran, M.,
Alfarhood, S., and Mridha, M. (2024). A multimodal
approach to cross-lingual sentiment analysis with en-
semble of transformer and llm. Scientific Reports,
14(1):9603.
Murray, C. J., Ikuta, K. S., Sharara, F., Swetschinski, L.,
Aguilar, G. R., Gray, A., Han, C., Bisignano, C., Rao,
P., Wool, E., et al. (2022). Global burden of bacterial
antimicrobial resistance in 2019: a systematic analy-
sis. The lancet, 399(10325):629–655.
Pehrsson, E. C., Tsukayama, P., Patel, S., Mej
´
ıa-Bautista,
M., Sosa-Soto, G., Navarrete, K. M., Calderon,
M., Cabrera, L., Hoyos-Arango, W., Bertoli, M. T.,
et al. (2016). Interconnected microbiomes and re-
sistomes in low-income human habitats. Nature,
533(7602):212–216.
Pham, V. H. and Kim, J. (2012). Cultivation of unculturable
soil bacteria. Trends in biotechnology, 30(9):475–484.
Rao, R. M., Liu, J., Verkuil, R., Meier, J., Canny, J. F.,
Abbeel, P., Sercu, T., and Rives, A. (2021). Trans-
former protein language models are unsupervised
structure learners. bioRxiv.
Rupp
´
e, E., Ghozlane, A., Tap, J., Pons, N., Alvarez, A.-S.,
Maziers, N., Cuesta, T., Hernando-Amado, S., Clares,
I., Mart
´
ınez, J. L., et al. (2019). Prediction of the
intestinal resistome by a three-dimensional structure-
based method. Nature microbiology, 4(1):112–123.
Steinegger, M. and S
¨
oding, J. (2018). Clustering huge pro-
tein sequence sets in linear time. Nature communica-
tions, 9(1):2542.
Suzek, B. E., Wang, Y., Huang, H., McGarvey, P. B.,
Wu, C. H., and Consortium, U. (2015). Uniref clus-
ters: a comprehensive and scalable alternative for im-
proving sequence similarity searches. Bioinformatics,
31(6):926–932.
Antibiotic Resistance Gene Identification from Metagenomic Data Using Ensemble of Finetuned Large Language Models
111

Taylor, W. L. (1953). “cloze procedure”: A new tool
for measuring readability. Journalism quarterly,
30(4):415–433.
Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones,
L., Gomez, A. N., Kaiser, Ł., and Polosukhin, I.
(2017). Attention is all you need. Advances in neural
information processing systems, 30.
Wang, Z., Li, S., You, R., Zhu, S., Zhou, X. J., and
Sun, F. (2021). Arg-shine: improve antibiotic resis-
tance class prediction by integrating sequence homol-
ogy, functional information and deep convolutional
neural network. NAR Genomics and Bioinformatics,
3(3):lqab066.
Wu, J., Ouyang, J., Qin, H., Zhou, J., Roberts, R., Siam, R.,
Wang, L., Tong, W., Liu, Z., and Shi, T. (2023). Plm-
arg: antibiotic resistance gene identification using a
pretrained protein language model. Bioinformatics,
39(11):btad690.
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M.,
Rasmussen, S., Lund, O., Aarestrup, F. M., and
Larsen, M. V. (2012). Identification of acquired an-
timicrobial resistance genes. Journal of antimicrobial
chemotherapy, 67(11):2640–2644.
KDIR 2024 - 16th International Conference on Knowledge Discovery and Information Retrieval
112
