
Improving Antibody-Antigen Interaction Prediction Through Flexibility
with ESMFold
Sara Joubbi
1,2 a
, Giuseppe Maccari
2 b
, Giorgio Ciano
2 c
, Alessio Micheli
1 d
,
Paolo Milazzo
1 e
and Duccio Medini
2 f
1
Department of Computer Science, University of Pisa, Pisa, Italy
2
Data Science for Health Lab, Fondazione Toscana Life Sciences, Siena, Italy
Keywords:
Antibody, Antigen, Fingerprint, Deep Learning, Flexibility, ESMFold.
Abstract:
Antibodies are essential proteins in the immune system due to their capacity to bind to specific antigens.
They also play a critical role in developing vaccines and treatments for infectious diseases. Their complex
structure, with variable regions for antigen binding and flexible hinge regions, presents challenges for ac-
curate computational modeling. Recent advancements in deep learning have revolutionized protein structure
prediction. Despite these advancements, predicting interactions between antibodies and antigens remains chal-
lenging, mainly due to the flexibility of antibodies and the dynamic nature of binding events. This study uses
fingerprint-based methodologies that incorporate ESMFold confidence scores as a flexibility feature to model
Ab-Ag interactions. Our methodology shows how including flexibility has improved Ab-Ag interactions by
3%, achieving an AUC-ROC of 91%.
1 INTRODUCTION
Antibodies are essential immune system proteins, re-
sponsible for identifying and binding to specific anti-
gens, such as pathogens or foreign substances, to neu-
tralize or mark them for destruction by other immune
cells (Kindt et al., 2007). This highly selective bind-
ing ability plays a critical role in immune defense and
makes antibodies invaluable tools in biotherapeutics.
They are widely used in developing treatments for
various diseases, including cancers, autoimmune dis-
orders, and infectious diseases, where they can target
specific molecules or cells with precision, minimizing
damage to healthy tissues (Kaplon et al., 2023).
Structurally, antibodies are Y-shaped proteins
composed of two heavy (H) and light (L) chains. Each
pair forms a variable (V) region that binds antigens,
while the constant (C) region, held together by disul-
fide bonds, binds to receptors and maintains protein
integrity (Joubbi et al., 2024) (see Figure 1A). Anti-
a
https://orcid.org/0009-0002-1079-7204
b
https://orcid.org/0000-0002-2894-3583
c
https://orcid.org/0000-0003-2863-4315
d
https://orcid.org/0000-0001-5764-5238
e
https://orcid.org/0000-0002-7309-6424
f
https://orcid.org/0000-0001-6041-2603
bodies present flexible hinge regions that connect the
antigen-binding fragment (Fab) to the crystallizable
fragment (Fc), enabling dynamic movement between
these regions. This flexibility allows the antibody
to better interact with various antigens and immune
receptors. Additionally, post-translational modifica-
tions, like glycosylation, play a critical role in reg-
ulating the antibody’s structure and function. Gly-
cosylation can influence the antibody’s stability, im-
mune recognition, and effector functions, contribut-
ing to its overall complexity and adaptability in im-
mune responses (Guo et al., 2024).
The field of antibody-based treatments is grow-
ing rapidly, as shown by the increasing number of
FDA approvals, clinical trials, and patent applications
(Wilman et al., 2022). The market for antibody thera-
pies is projected to surpass $400 billion by 2028, with
an annual growth rate of 14.1% (Larrosa et al., 2023;
Joubbi et al., 2024). Traditionally, antibody devel-
opment depends on labor-intensive and costly tech-
niques such as phage display and animal immuniza-
tion. However, the incorporation of computational
tools in pharmaceutical research is expected to greatly
reduce the costs and time associated with developing
new antibodies. This progress is expected to make
immunotherapy more affordable and suitable for a
broader range of diseases.
Joubbi, S., Maccari, G., Ciano, G., Micheli, A., Milazzo, P. and Medini, D.
Improving Antibody-Antigen Interaction Prediction Through Flexibility with ESMFold.
DOI: 10.5220/0013189500003911
Paper published under CC license (CC BY-NC-ND 4.0)
In Proceedings of the 18th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2025) - Volume 1, pages 603-610
ISBN: 978-989-758-731-3; ISSN: 2184-4305
Proceedings Copyright © 2025 by SCITEPRESS – Science and Technology Publications, Lda.
603
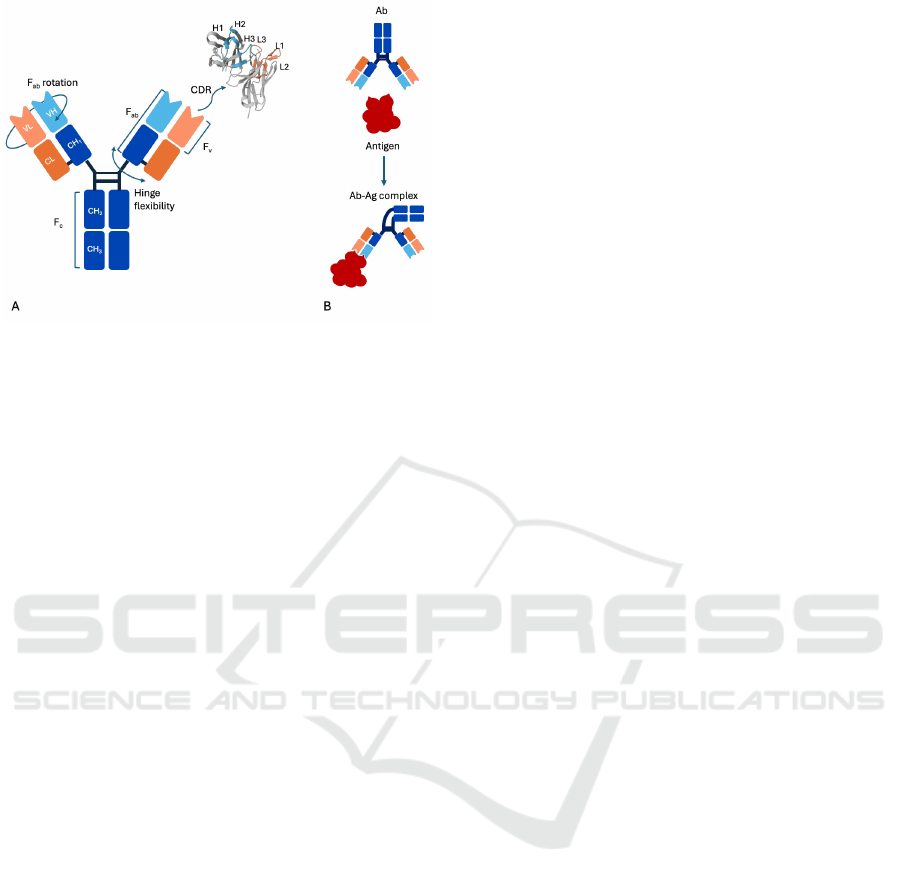
Figure 1: Antibody structure and flexibility. A) The heavy
chain (H) of the antibody is shown in blue, while the light
chain (L) is depicted in orange. On the right, a focus on
a CDR is shown with labeled light and heavy chain CDR
loops (PDB 3IY3). The Fab region is formed by the variable
regions and part of the constant (C) regions. The variable
regions (VH and VL) form the Fv region. The Fc region
contains the constant part of the chain. The antibody is a
flexible molecule due to the Fab rotation and Hinge flexibil-
ity. B) An example of antibody flexibility after binding to
an antigen. Additionally, there is a change in conformation
at the binding site with the antigen. The antibody can then
form different Fc-Fc configurations with another antibody
or bind with a cell receptor.
Deep Learning (DL) methods have addressed var-
ious biological challenges, notably with AlphaFold2
(AF2) (Jumper et al., 2021), which have transformed
structural biology by accurately predicting protein 3D
structures from amino acid sequences. AF2, despite
its success, relies on multiple sequence alignments
(MSAs), which are less effective for antibody fold-
ing due to the high variability and lack of evolution-
ary data in CDR H3 loop sequences (Joubbi et al.,
2024). Alternative methods have been developed to
overcome this limitation. ESMFold uses ESM-2 for
comprehensive embedded representations of protein
sequences, providing a viable alternative to MSAs
(Lin et al., 2023). ESMFold outperforms AF2 when
utilizing only the amino acid sequence, achieving a
TM-Score of 0.68 compared to AF2’s 0.37, while also
providing faster predictions (Bertoline et al., 2023).
Accurate prediction of paratope and epitope re-
gions is crucial for antibody design. While antibody-
antigen (Ab-Ag) interactions are a type of protein-
protein interaction (PPI), they have distinct char-
acteristics that make general PPI prediction meth-
ods less effective for antibody applications (Graves
et al., 2020). Several DL methods have been de-
veloped to address PPIs, including fingerprint (sur-
face) methods such as MaSIF (Gainza et al., 2020)
and dMaSIF (Sverrisson et al., 2021), as well as
PeSTo (Krapp et al., 2023). Additionally, specific
methods have been created for antibodies, such as
EMPM (Del Vecchio et al., 2021), PECAN (Pit-
tala and Bailey-Kellogg, 2020), and fingerprint-based
techniques like Surface ID (Riahi et al., 2023).
Future directions for modeling antibody-antigen
(Ab-Ag) interactions involve representing antibody
flexibility (Guo et al., 2024; Rudden et al., 2022;
Joubbi et al., 2024), as the paratope is characterized
by a certain level of flexibility (Wang et al., 2013;
Rosen et al., 2005) as shown in Figure 1B. More-
over, the two protein structures slightly change during
binding (Pegoraro et al., 2023). While DL networks
can incorporate local flexibility, they often struggle
with conformational switching (Rudden et al., 2022).
Addressing this remains a challenge, as long molecu-
lar dynamics simulations are computationally inten-
sive, and simpler analytical models, though faster,
may lack detail. Combining local contact models with
predicted Local Distance Difference Test (pLDDT)
scores can predict protein flexibility faster (Ma et al.,
2023; Alderson et al., 2023; Alderson et al., 2023;
Middendorf and Eicholt, 2024). Building on this
approach, we use ESMFold’s pLDDT as a flexibil-
ity feature for Ab-Ag interactions using fingerprint
methodologies.
Main Contributions:
1. Application of the dMaSIF model to antibody-
antigen (Ab-Ag) interactions, incorporating pro-
tein flexibility into the analysis.
2. Utilization of pLDDT scores to estimate the flex-
ibility of Ab-Ag interactions, demonstrating the
potential for performance enhancement.
2 RELATED WORKS
Fischer in 1894 discovered that the interactions be-
tween molecules are heavily influenced by their struc-
ture and arrangement and successful binding relies
on the compatibility of geometric shapes (Fischer,
1894). Following this concept, several DL meth-
ods that target PPI and Ab-Ag interaction are based
on the structure and surface of the protein. PeSTo
(Krapp et al., 2023) is a revolutionary parameter-free
geometric transformer that directly manipulates the
atomic components of a protein structure. This in-
novative approach accurately predicts specific regions
on a protein surface that have the potential to in-
teract with other proteins, as well as nucleic acids,
lipids, ions, and small molecules. MaSIF (Gainza
et al., 2020) is another pioneering method that em-
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
604

ploys DL and the concept of fingerprints to forecast
PPIs. It accomplishes this by creating protein finger-
prints based on amino acid sequences, structural el-
ements, and functional motifs. The method divides
protein surfaces into patches and utilizes a convolu-
tional neural network (CNN) to identify interaction
sites and patterns. MaSIF has diverse applications, in-
cluding ligand binding (MaSIF-ligand), interface site
prediction (MaSIF-site), and partner binding predic-
tion (MaSIF-search). However, MaSIF’s reliance on
pre-computed features and meshes leads to slow per-
formance and high memory usage. To tackle these
issues, dMaSIF (Sverrisson et al., 2021) operates di-
rectly on raw 3D coordinates and atom types. It gen-
erates molecular surfaces on the fly using a novel ge-
ometric convolutional layer, making it significantly
faster and more memory-efficient than MaSIF. Sur-
face ID (Riahi et al., 2023) employs MaSIF for Ab-
Ag interaction predictions.
Additional Ab-Ag-specific methods have been de-
veloped, such as PECAN (Pittala and Bailey-Kellogg,
2020), which uses a symmetrical graph convolutional
network (GCN) to predict both paratopes and epitopes
within a unified framework, and EPMP (Del Vec-
chio et al., 2021), which separates the prediction
models for paratopes and epitopes. GEP (geometric
epitope–paratope) prediction (Pegoraro et al., 2023)
proposes geometric representations of molecules to
create accurate predictors for predicting antibody-
antigen binding sites. The study demonstrates the
significance of the surface in this type of interaction
and the usefulness of different geometric represen-
tation information for various tasks. Surface-based
models (OGEP) are more efficient in predicting epi-
tope binding, while graph models (IGEP) are better
for paratope prediction, resulting in significant perfor-
mance improvements. However, none of these meth-
ods take into account the binding’s flexibility, which
is a crucial factor to consider.
3 MATERIALS AND METHODS
In this section, we present the dataset used for this
study (Subsection 3.1), followed by an introduction
to the fingerprint method (Subsection 3.2) based on
dMaSIF and how we obtained and integrated flexi-
bility within the model (Subsection 3.3). Before ex-
amining the Ab-Ag interaction, we conducted a pre-
liminary comparison of the performance of PPI and
Ab-Ag interactions.
Table 1: Dataset composition.
Dataset Training Validation Test
PPI 4,449 494 959
Ab-Ag 2,729 303 535
3.1 Dataset
For the PPI task, we used the dMaSIF dataset (Sver-
risson et al., 2021). The dataset contains 4,943
protein-protein complexes used for training and val-
idation (10%), with an additional 959 complexes re-
served for testing. In the case of antibody-antigen
interactions, we downloaded a total of 16,269 Ab-
Ag complexes from the SAbDab database (Dunbar
et al., 2014) (April 2024). We then filtered nanobod-
ies, non-defined antigens, haptens, and non-protein
targets. Furthermore, we excluded structures with a
resolution lower than 4
˚
A. To evaluate the similarity of
antibody structures, we utilized the TM-Score (Zhang
and Skolnick, 2004) and excluded Ab-Ag complexes
where the antibody exhibited a TM-Score ¡ 30%. The
dataset was randomly split, resulting in 3,032 Ab-Ag
complexes for training and validation (10%), and 535
complexes for testing. We conducted hyperparameter
tuning and initial configuration study using the vali-
dation set. A summary of the dataset composition is
presented in Table 1.
3.2 Method
Our method is based on dMaSIF (Sverrisson et al.,
2021), an efficient end-to-end geometric analysis ar-
chitecture. The underlying idea is that geometric and
chemical features provide crucial information about
the protein’s surface, especially for PPIs. The method
overview is shown in Figure 2. The model samples
the surface points and normals of the protein and
proceeds to compute mean and Gaussian curvatures
at multiple scales (Figure 2b and c). Chemical fea-
tures are derived based on atom types and their in-
verse distances to surface points and are processed
through a multi-layer perceptron (MLP) (Figure 2d).
These chemical and curvature features form a 16-
dimensional feature vector (Figure 2e). An MLP is
then utilized to predict orientation scores for each sur-
face point, which are employed to align local coor-
dinates (Figure 2f). Further, trainable convolutions
and MLPs refine the feature vectors (Figure 2g), and
interaction prediction is executed by calculating dot
products between the feature vectors of two proteins
to generate interaction scores (Figure 2h). In the fol-
lowing subsections, we describe how we have added
the concept of flexibility to this process. In Figure 3,
there is a summary of the changes made to the original
model to include flexibility and interactive flexibility.
Improving Antibody-Antigen Interaction Prediction Through Flexibility with ESMFold
605
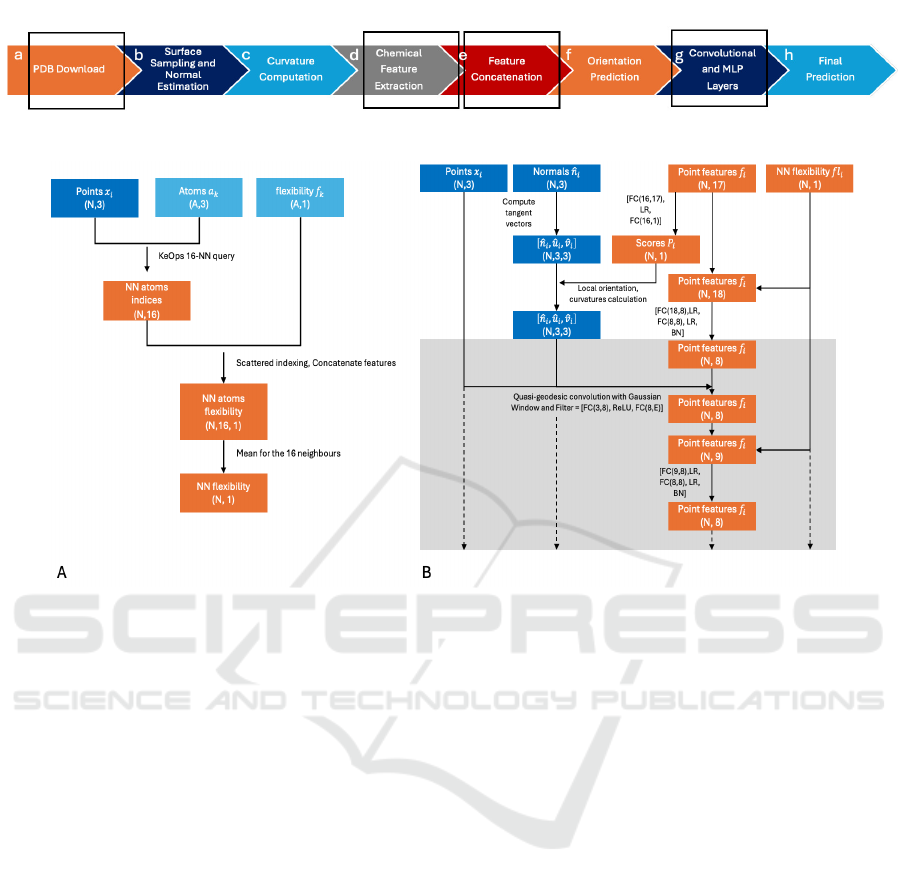
Figure 2: Overview of the dMaSIF method. The red squares represent areas where flexibility was added.
Figure 3: A) This diagram illustrates how flexibility can be represented on the surface of the protein. For each point on the
surface, we computed the 16 nearest neighbors of the corresponding atom in the structure and assigned a flexibility score to
each of the N points, calculated as the mean value of the flexibility scores of the 16 neighbors. B) Architecture of iterative
flexibility. In comparison to the original dMaSIF model, we incorporated the right block of flexibility as an additional feature
in each MLP block, both before and after the quasi-geodesic convolution. The gray box represents one layer of the network,
which can be repeated to create multiple layers prior to generating the output embeddings of the model.
3.2.1 Data Representation
Each protein is represented by a 3D point cloud that
captures every atom in the protein. Following the
dMaSIF representation, each atom is characterized by
10 geometric features (5+5 mean and Gaussian cur-
vatures) and 6 chemical features (one-hot encoding of
the six most significant atoms: C, H, O, N, S, Se).
Additionally, we incorporated the pLDDT score from
ESMFold to indicate residue flexibility. The pLDDT
score ranges from 0 to 100, with higher scores indi-
cating greater certainty in the model’s predictions re-
garding the atom’s folding, while lower scores sug-
gest increased uncertainty in the final folding, as il-
lustrated in Figure 4A. Studies have demonstrated
that this score correlates with protein flexibility (Guo
et al., 2024; Rudden et al., 2022; Ma et al., 2023):
higher scores correspond to lower flexibility and vice
versa. The original dMaSIF representation, without
flexibility, had 16 features, whereas our method in-
cludes 17 features.
3.3 Flexibility Score with ESMFold
ESMFold (Lin et al., 2023) uses ESM2, which offers
a comprehensive embedded representation of protein
sequences (Joubbi et al., 2024). At the end of the fold-
ing process, this method generates the pLDDT score
mentioned above. To obtain this score, we folded the
corresponding sequences and generated a PDB file.
In the resulting PDB file, the pLDDT score is saved
as the b-factor. One issue we encountered was that
dMaSIF uses protonated structures, whereas the final
PDB files generated by ESMFold do not include hy-
drogen scores. To address this, we created a flexibility
score for each residue instead of individual atoms. In
the end, each atom has a flexibility score correspond-
ing to the residue flexibility, as shown in Figure 4B.
3.3.1 3D Point Cloud Association with the
Flexibility Score
dMaSIF samples some point on the surface and com-
putes the Gaussian curvatures, then the chemical fea-
tures are computed based on atom types and their
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
606

Figure 4: A) Example of a prediction using ESMFold. Given an input sequence, ESMFold predicts the three-dimensional
structure, with the pLDDT score reflecting the model’s confidence for each structural atom. On the left is the original protein
structure, while the right displays the predicted structure alongside the pLDDT score. The protein structure is PDB 1L8W. B)
Assignment of features for each protein structure. Each atom possesses geometric and chemical features, while flexibility is
uniform across all atoms within a single residue.
inverse distances to surface points. These features
are then processed through a multi-layer perceptron
(MLP) with six hidden units, ReLU activation, and
batch normalization. In our model, to report the flexi-
bility on the protein’s surface, as shown in Figure 3A,
we only performed a 16 nearest-neighbor search since
ESMFold has already pre-processed this feature. We
took the average flexibility score of these 16 neigh-
bors for each point. Finally, these features are con-
catenated into a vector of 17 elements.
3.3.2 dMaSIF Network Modifications
The network is based on trainable convolutions,
MLPs, and batch normalization for the feature vec-
tors. We evaluated two options for incorporating the
flexibility feature: using it directly or employing iter-
ative flexibility layers. We chose iterative layers be-
cause flexibility accounts for only 1/17 of the features,
and interactively adding it helps amplify its signifi-
cance. This adjustment resulted in a feature vector
with 18 elements, as shown in Figure 3B. For inter-
action prediction, dot products are computed between
the feature vectors of both proteins to generate inter-
action scores.
Figure 5: Ablation study for the different combinations of
features. ”geom” denotes geometrical features, ”chem” rep-
resents chemical features, and ”flex” indicates flexibility.
The different models are the following: ”PPI flex” = model
trained on PPI data using flexibility; ”PPI iter” = model
trained on PPI data using iterative flexibility; ”PPI flex
(Ab)” = inference on Ab-Ag complexes using the model
trained on PPI + flexibility, ”PPI iter (Ab)” = inference on
Ab-Ag complexes using the model trained on PPI + iterative
flexibility; ”Ab-Ag fine flex” = model fine-tuned on Ab-Ag
complex + flexibility; ”Ab-Ag fine iter” = model fine-tuned
on Ab-Ag complex + iterative flexibility; ”Ab-Ag flex” =
model trained from scratch on Ab-Ag complexes + flexibil-
ity; ”Ab-Ag iter” = model trained from scratch on Ab-Ag
complexes + iterative flexibility.
Improving Antibody-Antigen Interaction Prediction Through Flexibility with ESMFold
607

Table 2: Results are presented in terms of ROC-AUC for the 5-fold cross-validation on the test set. The first three columns
represent the model trained on PPI (PPI), the inference on Ab-Ag (Ab-Ag inference), and the fine-tuning on Ab-Ag (Ab-Ag
fine-tuning). The final column indicates the model trained from scratch using Ab-Ag structures (Ab-Ag). The ’Original’ row
represents the model without flexibility, the ’Flexibility’ row denotes the model with the flexibility feature, and the ’Iterative
Flexibility’ row presents the results for the iterative model with flexibility. The best result from each experiment is highlighted
in bold.
Model PPI
Ab-Ag
inference
Ab-Ag
fine-tuning
Ab-Ag
Original 0.835±0.002 0.832±0.004 0.898±0.004 0.881±0.004
Flexibility 0.783±0.003 0.765±0.017 0.863±0.006 0.895±0.002
Iterative
flexibility
0.778±0.008 0.765±0.011 0.866±0.002 0.910±0.002
4 RESULTS
4.1 Cross-Validation
As an initial approach to the Ab-Ag interaction prob-
lem, we assessed whether the dMaSIF model, both
with and without flexibility, could effectively gener-
alize to the Ab-Ag interaction task. We trained the
dMaSIF model using PPI data, as this dataset has
been successfully utilized in prior applications of the
model. As indicated in Table 2, the inclusion of flex-
ibility in the PPI data did not improve predictions
both for PPI and Ab-Ag interaction tasks. In a sub-
sequent attempt, we fine-tuned the PPI-trained model
using Ab-Ag data, which resulted in improved per-
formance; however, the model with flexibility did not
outperform the one without it. Ultimately, we trained
the model from scratch using Ab-Ag data, and in
this scenario, the incorporation of flexibility signifi-
cantly enhanced performance. These results demon-
strate that Ab-Ag interactions represent a specific cat-
egory of PPI and highlight the necessity for a dedi-
cated model to better characterize them, as well as the
benefits of including flexibility to enhance results. Ta-
ble 2 shows the results of the 5-fold cross-validation.
4.2 Comparison with OGEP
We compared our model with OGEP, the leading
benchmark method for analyzing antibody-antigen in-
teractions based on surface data. We used the GEP
test set, excluding any Protein Data Bank (PDB) en-
tries that overlapped with our training data to en-
sure a fair comparison. This filtering process re-
sulted in a test set of 29 unique PDB entries used
with Ab-Ag iterative flexibility. Although the com-
parison is primarily indicative due to the limited data
available, our findings indicate that our model, which
was trained on Ab-Ag from scratch using iterative
flexibility, demonstrates significantly superior perfor-
mance compared to OGEP (PINet) for antigen in-
teractions (OGEP AUC-ROC: 0.77±0.03 vs. Ab-
Ag iterative AUC-ROC: 0.97±0.00). Additionally, it
achieves comparable performance for antibody inter-
actions (OGEP AUC-ROC: 0.77 ± 0.02 vs. Ab-Ag
iterative AUC-ROC: 0.75 ± 0.01).
4.3 Impact of the Different Features on
Final Prediction
In this work, we conducted an ablation study focusing
on various model features to evaluate the importance
of flexibility in protein-protein interactions (PPI) and
antibody-antigen (Ab-Ag) interactions. We analyzed
all possible combinations of three primary feature
groups: geometrical, chemical, and flexibility. As il-
lustrated in Figure 5, all methods are influenced by
both chemical and flexibility features, although their
dependence on individual features varies. The PPI in-
teraction models place a greater emphasis on chemi-
cal attributes. Conversely, the fine-tuning model sug-
gests that flexibility features alone have become in-
creasingly important for predictions, except for the
model utilizing iterative flexibility, where geometri-
cal features assume a more significant role. Similar
trends were observed in the Ab-Ag interaction model
trained from scratch, highlighting that pLDDT pro-
vides robust predictive features. This highlights the
critical role of the flexibility score in Ab-Ag interac-
tions.
5 CONCLUSIONS
Antibody-antigen interactions are critical molecu-
lar events forming the basis for immune recogni-
tion and neutralization of pathogens or foreign sub-
stance. While different computational approaches has
been developed to model antibody-antigen interac-
tion, most overlook protein flexibility. By incorporat-
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
608

ing pLDDT scores from ESMFold as a proxy for flex-
ibility, we demonstrated a 3% improvement in predic-
tion accuracy, achieving an AUC-ROC of 91%. No-
tably, models that explicitly prioritized flexibility out-
performed those that considered flexibility to a lesser
extent, highlighting its significance in enhancing pre-
dictive capabilities. While this represents an initial ef-
fort to integrate flexibility into antibody-antigen mod-
eling, future approaches could utilize experimentally
derived configurations of antibody-antigen complexes
or energy-based models to simulate this dynamic be-
havior more effectively.
A key limitation of the current method is its re-
liance on pre-processed pLDDT scores, which in-
troduces computational overhead. To address this,
we propose incorporating structural distillation tech-
niques to embed flexibility-related insights directly
into sequence-based models, thereby eliminating the
need for structural preprocessing. This adaptation
would streamline workflow and enhance accessibility
for experimental laboratories by enabling rapid high-
throughput screening of antibody libraries.
In practical terms, this methodology holds
promise for applications such as epitope mapping
and evaluating binding interactions. By identifying
promising antibody candidates earlier in the process,
researchers can concentrate experimental resources
on the most viable options, accelerating the develop-
ment of effective antibody therapies.
DATA AVAILABILITY
The data and code can be accessed at the following
link: https://github.com/dasch-lab/fingerprint.
ACKNOWLEDGEMENTS
This work is partially supported by PNRR
ECS00000017 Tuscany Health Ecosystem - Spoke
6 ”Precision medicine & personalized healthcare”,
funded by the European Commission under the
NextGeneration EU program. We thank the Univer-
sity of Pisa Data Center for providing the necessary
hardware resources for this project. We thank Pietro
Li
`
o and St
´
ephane M. Gagn
´
e for their valuable
feedback.
REFERENCES
Alderson, T. R., Priti
ˇ
sanac, I., Kolari
´
c, Moses, A. M.,
and Forman-Kay, J. D. (2023). Systematic identifi-
cation of conditionally folded intrinsically disordered
regions by alphafold2. Proceedings of the National
Academy of Sciences, 120(44):e2304302120.
Bertoline, L. M., Lima, A. N., Krieger, J. E., and Teix-
eira, S. K. (2023). Before and after alphafold2: An
overview of protein structure prediction. Frontiers in
bioinformatics, 3:1120370.
Del Vecchio, A., Deac, A., Li
`
o, P., and Veli
ˇ
ckovi
´
c, P.
(2021). Neural message passing for joint paratope-
epitope prediction. arXiv preprint arXiv:2106.00757.
Dunbar, J., Krawczyk, K., Leem, J., Baker, T., Fuchs,
A., Georges, G., et al. (2014). Sabdab: the struc-
tural antibody database. Nucleic Acids Research,
42(D1):D1140–D1146.
Fischer, E. (1894). Einfluss der configuration auf die
wirkung der enzyme. Berichte der deutschen chemis-
chen Gesellschaft, 27(3):2985–2993.
Gainza, P., Sverrisson, F., Monti, F., Rodola, E., Boscaini,
D., Bronstein, M., and Correia, B. (2020). Deci-
phering interaction fingerprints from protein molec-
ular surfaces using geometric deep learning. Nature
Methods, 17(2):184–192.
Graves, J., Byerly, J., Priego, E., Makkapati, N., Parish,
S. V., Medellin, B., and Berrondo, M. (2020). A re-
view of deep learning methods for antibodies. Anti-
bodies, 9(2):12.
Guo, D., De Sciscio, M. L., Ng, J. C.-F., and Fraternali,
F. (2024). Modelling the assembly and flexibility of
antibody structures. Current Opinion in Structural Bi-
ology, 84:102757.
Joubbi, S., Micheli, A., Milazzo, P., Maccari, G., Ciano, G.,
Cardamone, D., and Medini, D. (2024). Antibody de-
sign using deep learning: from sequence and structure
design to affinity maturation. Briefings in Bioinfor-
matics, 25(4):bbae307.
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov,
M., Ronneberger, O., Tunyasuvunakool, K., Bates, R.,
ˇ
Z
´
ıdek, A., Potapenko, A., et al. (2021). Highly accu-
rate protein structure prediction with alphafold. Na-
ture, 596(7873):583–589.
Kaplon, H., Crescioli, S., Chenoweth, A., Visweswaraiah,
J., and Reichert, J. M. (2023). Antibodies to watch in
2023. In MAbs, volume 15, page 2153410. Taylor &
Francis.
Kindt, T. J., Goldsby, R. A., Osborne, B. A., and Kuby, J.
(2007). Kuby immunology. Macmillan.
Krapp, L. F., Abriata, L. A., Cort
´
es Rodriguez, F., and
Dal Peraro, M. (2023). Pesto: parameter-free geo-
metric deep learning for accurate prediction of pro-
tein binding interfaces. Nature communications,
14(1):2175.
Larrosa, C., Mora, J., and Cheung, N.-K. (2023). Global
impact of monoclonal antibodies (mabs) in children:
a focus on anti-gd2. Cancers, 15(14):3729.
Lin, Z., Akin, H., Rao, R., Hie, B., Zhu, Z., Lu, W.,
Smetanin, N., Verkuil, R., Kabeli, O., Shmueli, Y.,
et al. (2023). Evolutionary-scale prediction of atomic-
level protein structure with a language model. Sci-
ence, 379(6637):1123–1130.
Improving Antibody-Antigen Interaction Prediction Through Flexibility with ESMFold
609

Ma, P., Li, D.-W., and Br
¨
uschweiler, R. (2023). Predicting
protein flexibility with alphafold. Proteins: Structure,
Function, and Bioinformatics, 91(6):847–855.
Middendorf, L. and Eicholt, L. A. (2024). Random, de
novo, and conserved proteins: how structure and dis-
order predictors perform differently. Proteins: Struc-
ture, Function, and Bioinformatics, 92(6):757–767.
Paszke, A., Gross, S., Massa, F., Lerer, A., Bradbury, J.,
Chanan, G., et al. (2019). Pytorch: An imperative
style, high-performance deep learning library. Ad-
vances in Neural Information Processing Systems, 32.
Pegoraro, M., Domin
´
e, C., Rodol
`
a, E., Veli
ˇ
ckovi
´
c, P., and
Deac, A. (2023). Geometric epitope and paratope pre-
diction. bioRxiv.
Pittala, S. and Bailey-Kellogg, C. (2020). Learning context-
aware structural representations to predict antigen
and antibody binding interfaces. Bioinformatics,
36(13):3996–4003.
Riahi, S., Lee, J. H., Sorenson, T., Wei, S., Jager, S., Olfati-
Saber, R., Zhou, Y., Park, A., Wendt, M., Minoux, H.,
et al. (2023). Surface id: a geometry-aware system for
protein molecular surface comparison. Bioinformat-
ics, 39(4):btad196.
Rosen, O., Chill, J., Sharon, M., Kessler, N., Mester, B.,
Zolla-Pazner, S., and Anglister, J. (2005). Induced fit
in hiv-neutralizing antibody complexes: evidence for
alternative conformations of the gp120 v3 loop and
the molecular basis for broad neutralization. Biochem-
istry, 44(19):7250–7258.
Rudden, L. S., Hijazi, M., and Barth, P. (2022). Deep
learning approaches for conformational flexibility and
switching properties in protein design. Frontiers in
Molecular Biosciences, 9:928534.
Sverrisson, F., Feydy, J., Correia, B. E., and Bronstein,
M. M. (2021). Fast end-to-end learning on protein sur-
faces. In Proceedings of the IEEE/CVF Conference
on Computer Vision and Pattern Recognition, pages
15272–15281.
Wang, W., Ye, W., Yu, Q., Jiang, C., Zhang, J., Luo, R.,
and Chen, H.-F. (2013). Conformational selection and
induced fit in specific antibody and antigen recogni-
tion: Spe7 as a case study. The journal of physical
chemistry B, 117(17):4912–4923.
Wilman, W., Wr
´
obel, S., Bielska, W., Deszynski,
P., Dudzic, P., Jaszczyszyn, I., Kaniewski, J.,
Młokosiewicz, J., Rouyan, A., Satława, T., et al.
(2022). Machine-designed biotherapeutics: opportu-
nities, feasibility and advantages of deep learning in
computational antibody discovery. Briefings in Bioin-
formatics, 23(4):bbac267.
Zhang, Y. and Skolnick, J. (2004). Scoring function for au-
tomated assessment of protein structure template qual-
ity. Proteins: Structure, Function, and Bioinformat-
ics, 57(4):702–710.
APPENDIX
Environment Settings
The hardware and software resources used are pre-
sented in Table 3.
Table 3: Development environments and requirements.
System Ubuntu 20.04.5 LTS
CPU AMD EPYC 7413 24-Core Pro-
cessor
RAM 16×4GB; 2.67MT/s
GPU NVIDIA A100-SXM-80GB
CUDA
version
11.5
Programming
language
Python 3.8.18
Deep learning
framework
Pytorch (Paszke et al., 2019)
(Torch 1.12.1, torchvision
0.13.1, torchaudio 0.12.1)
Model Training and Hyperparameters
For dMaSIF pre-training on protein-protein interac-
tions (PPI) and fine-tuning on antibody-antigen (Ab-
Ag) interactions, we used a 9.0 radius, 8 embedding
dimensions, and one layer. For Ab-Ag models trained
from scratch, the non-flexibility version used the same
parameters as dMaSIF, while the flexibility-enhanced
models used a 10.0 radius, 16 embedding dimensions,
and either 3 layers (non-iterative) or 5 layers (itera-
tive). All models were trained with a batch size of 8
for 50 epochs, utilizing early stopping, binary cross-
entropy loss, and AMSGrad with a learning rate of
3e-4.
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
610
