
Machine Learning-Based Prediction of Key Genes Correlated to the
Subretinal Lesion Severity in a Mouse Model of Age-Related Macular
Degeneration
Kuan Yan
1
, Yue Zeng
2,3
, Dai Shi
1
, Ting Zhang
2
, Dmytro Matsypura
1
, Mark C. Gillies
2
, Ling Zhu
2,∗
and Junbin Gao
1,∗
1
Discipline of Business Analytics, Business School, The University of Sydney, Camperdown, NSW 2006, Australia
2
Macula Research Group, Save Sight Institute, Faculty of Medicine and Health, The University of Sydney, Camperdown,
NSW 2006, Australia
3
Department of Ophthalmology, The First Affiliated Hospital of Zhejiang University, Hangzhou, Zhejiang Province, China
{kuan.yan, yue.zeng, dai.shi, ting.zhang, dmytro.matsypura, mark.gillies, ling.zhu, junbin.gao}@sydney.edu.au
Keywords:
Age-Related Macular Degeneration, Machine Learning, Subretinal Lesion Severity, Subretinal Fibrosis, RNA
Sequencing, Genetic Targets.
Abstract:
Age-related macular degeneration (AMD) is a major cause of blindness in older adults, severely affecting
vision and quality of life. Despite advances in understanding AMD, the molecular factors driving the sever-
ity of subretinal scarring (fibrosis) remain elusive, hampering the development of effective therapies. This
study introduces a machine learning-based framework to predict key genes that are strongly correlated with
lesion severity and to identify potential therapeutic targets to prevent subretinal fibrosis in AMD. Using an
original RNA sequencing (RNA-seq) dataset from the diseased retinas of JR5558 mice, we developed a novel
and specific feature engineering technique, including pathway-based dimensionality reduction and gene-based
feature expansion, to enhance prediction accuracy. Two iterative experiments were conducted by leveraging
Ridge and ElasticNet regression models to assess biological relevance and gene impact. The results highlight
the biological significance of several key genes and demonstrate the framework’s effectiveness in identifying
novel therapeutic targets. The key findings provide valuable insights for advancing drug discovery efforts and
improving treatment strategies for AMD, with the potential to enhance patient outcomes by targeting the un-
derlying genetic mechanisms of subretinal lesion development.
1 INTRODUCTION
Neovascular age-related macular degeneration
(nAMD) is the leading cause of blindness in indi-
viduals aged 50 and older (Blindness, 2021). While
anti-vascular endothelial growth factor (VEGF)
therapies have been the gold standard for treating
nAMD, long-term studies reveal that up to 70%
of patients treated with anti-VEGF drugs develop
subretinal fibrosis within 10 years, resulting in severe
visual loss (Gillies et al., 2020; Bloch et al., 2013).
Currently, no FDA-approved treatments exist for
subretinal fibrosis, making the identification of novel
genetic targets and biological pathways critical for
improving visual outcomes in nAMD.
The spontaneous JR5558 mouse model (Won
et al., 2011), developed at the Jackson Laboratory,
∗
Co-corresponding authors.
offers a valuable tool for studying the progression
of nAMD. These mice develop subretinal fibrovascu-
lar lesions, visible as yellow mounds in fundus pho-
tographs, starting at 4 weeks and expanding until 12
weeks of age. A critical angio-fibrotic switch oc-
curs at around 8 weeks, making the model particularly
suited for examining both early neovascular changes
and late-stage fibrosis (Nagai et al., 2014; Hasegawa
et al., 2014; Linder et al., 2024). Therapeutic tar-
gets for subretinal fibrosis have been validated in this
model (Rossato et al., 2020). However, traditional
methods of studying nAMD, which rely on observa-
tional and statistical techniques, often fall short due
to the complexity and vastness of genetic data. These
conventional approaches lack the precision and scal-
ability required to identify specific genes that drive
disease progression, underscoring the need for novel
strategies that can address these challenges.
Machine Learning (ML) offers significant poten-
Yan, K., Zeng, Y., Shi, D., Zhang, T., Matsypura, D., Gillies, M. C., Zhu, L. and Gao, J.
Machine Learning-Based Prediction of Key Genes Correlated to the Subretinal Lesion Severity in a Mouse Model of Age-Related Macular Degeneration.
DOI: 10.5220/0013245600003911
Paper published under CC license (CC BY-NC-ND 4.0)
In Proceedings of the 18th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2025) - Volume 1, pages 627-637
ISBN: 978-989-758-731-3; ISSN: 2184-4305
Proceedings Copyright © 2025 by SCITEPRESS – Science and Technology Publications, Lda.
627

tial in the field of genomics, with its ability to ana-
lyze vast datasets and uncover intricate patterns that
may not be apparent through conventional analysis
(Bostanci et al., 2023; Chandrasekhar and Peddakr-
ishna, 2023). By applying ML models to RNA se-
quencing (RNA-seq) data, researchers can predict le-
sion severity and identify key genes associated with
subretinal fibrosis. Consequently, using ML to fo-
cus on disease severity in the JR5558 mouse model
can further aid in high-throughput screening for novel
therapeutic targets in subretinal fibrosis, thereby ad-
vancing drug discovery efforts. However, RNA-seq
datasets can vary significantly across different appli-
cations, often facing challenges such as limited sam-
ple sizes and high dimensionality. Effectively pre-
processing these datasets, selecting the most suitable
models and designing appropriate prediction tasks
have become critical challenges.
To address the aforementioned issues, we investi-
gate a novel problem in this paper: how to utilize ML
models to predict lesion severity and identify poten-
tial therapeutic gene targets in subretinal fibrosis re-
lated to AMD using RNA-seq data from mice. First,
we collected and organized comprehensive RNA-seq
data from the retinas of JR5558 mice. We then pre-
processed this original RNA-seq dataset by reducing
dimensionality and expanding features before training
the ML models. We employed Ridge and ElasticNet
regression models for training and prediction. To ver-
ify the effectiveness of our proposed framework and
further analyze the biological impact, we conducted
two sets of iterative experiments on biological corre-
lation and gene impact measurement.
The main contributions of our research can be
summarized as follows:
• We collected and provided an original and com-
prehensive RNA-seq dataset from the retinas of
JR5558 mice, facilitating further research in sub-
retinal fibrosis.
• We applied ML models, specifically Ridge and
ElasticNet regression, to predict lesion severity
and identify key genes associated with subretinal
fibrosis, thereby enhancing precision in genetic
analysis.
• We tackled challenges related to limited sample
sizes and high dimensionality in RNA-seq data,
improving data preprocessing and model selection
strategies in transcriptomic research.
• We designed and conducted iterative experiments
based on the original datasets we collected and
produced, verifying the effectiveness and ex-
cellent performance of our proposed framework
through biological impact analysis in two dimen-
sions: biological correlation and gene impact
measurement.
• Our approach identifies potential therapeutic gene
targets, offering new insights into transcriptomic
influences on subretinal fibrosis and advancing
drug discovery efforts.
The remainder of this paper is structured as fol-
lows. Section 2 presents a literature review of ma-
chine learning in biomedical research, RNA-seq data
in machine learning and transcriptomic studies on dis-
ease severity and subretinal fibrosis. Section 3 il-
lustrates our proposed framework and methodology.
Section 4 details the dataset description, presents and
discusses the experimental results and biological im-
pact and Section 5 summarizes the paper’s conclu-
sions.
2 RELATED WORK
2.1 Machine Learning in Biomedical
Research
In recent years, the integration of ML into biomedical
research has revolutionized the field, offering novel
insights and predictive capabilities that were previ-
ously unattainable. As the volume of biomedical data
continues to grow exponentially, ML provides essen-
tial methods for analyzing intricate datasets, discov-
ering patterns and making informed predictions.
Traditional statistical methods, such as t-tests and
ANOVA, have been instrumental in biomedical re-
search but come with inherent limitations. These
methods often require assumptions about the data and
can struggle with the high dimensionality and com-
plexity typically for biological datasets. Furthermore,
traditional approaches may not always capture subtle
patterns and interactions within the data, leading to
potential oversights. In contrast, ML offers significant
advantages in handling large and complex datasets.
ML algorithms can uncover intricate patterns and re-
lationships without the need for predefined models,
enhancing the ability to make accurate predictions
and discoveries. This flexibility is particularly bene-
ficial in multi-omics studies, where the complexity of
the data demands more robust and adaptive analytical
techniques.
ML techniques have significantly advanced
biomedical research, enhancing our ability to address
various critical aspects such as predicting disease out-
comes, identifying biomarkers and uncovering thera-
peutic targets. In the realm of predicting disease out-
comes, Khan et al. (Khan et al., 2023) and Bhatt
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
628

et al. (Bhatt et al., 2023) focused on cardiovascu-
lar diseases, with Khan’s employing random forest
(RF) algorithms and Bhatt’s utilizing a multilayer per-
ceptron with cross-validation, both significantly en-
hancing early diagnosis precision. Arumugam et al.
(Arumugam et al., 2023) optimized a decision tree
model to predict heart disease in diabetes patients,
improving diagnostic accuracy and efficiency. Sim-
ilarly, Islam et al. (Islam et al., 2023) evaluated var-
ious ML models for chronic kidney disease predic-
tion, determining that the XGBoost classifier was the
most effective, thereby supporting timely intervention
and treatment planning. Transitioning to the identi-
fication of biomarkers, Zhang et al. (Zhang et al.,
2021) employed feature selection methods alongside
various ML techniques, including support vector ma-
chines (SVMs), to boost diagnostic accuracy and aid
in the development of targeted treatments. Mi et al.
(Mi et al., 2021) introduced PermFIT, a permutation-
based technique using RFs and SVMs, to identify cru-
cial biomarkers for complex diseases. This method
isolated key genetic markers, enhancing the under-
standing and diagnosis of intricate medical condi-
tions. In the quest to uncover therapeutic targets,
Pun et al. (Pun et al., 2023) utilized deep learning
models to analyze large-scale omics data, identifying
novel therapeutic targets for complex diseases by un-
covering critical molecular interactions and pathways.
Rafique et al. (Rafique et al., 2021) applied ensemble
learning methods, such as RFs and gradient boosting
machines, to predict patient responses to various can-
cer treatments. By analyzing clinical and molecular
data, they aimed to improve the accuracy of therapeu-
tic response predictions, ultimately enhancing patient
outcomes in oncology.
With continuous advancements in this field, ML
has demonstrated significant advantages in handling
large-scale biological datasets and uncovering mean-
ingful patterns. Consequently, it has become a valu-
able tool for processing and analyzing RNA-seq data.
2.2 RNA Sequencing Data in Machine
Learning
RNA-seq has significantly advanced biomedical re-
search and transcriptomics by offering a comprehen-
sive view of gene expression. Unlike traditional pro-
filing methods, RNA-seq quantifies transcript levels
across the entire genome, providing insights into gene
regulation and cellular functions (Slovin et al., 2021).
This detailed analysis enables the identification of
transcriptomic activity patterns associated with dis-
ease processes, which are crucial for understanding
complex biological systems. Additionally, it aids in
discovering potential biomarkers and therapeutic tar-
gets (Andrews et al., 2021). In ML applications, the
ability of RNA-seq data to capture the full range of
gene expression makes it an invaluable resource for
developing predictive models. These models can elu-
cidate the transcriptomic factors that influence disease
severity and progression.
RNA-seq data have been instrumental in uncover-
ing critical insights across a range of conditions, ulti-
mately contributing to improved diagnosis, treatment
and understanding of complex diseases. For instance,
in oncology, RNA-seq has been used to classify can-
cer subtypes that predict cancer progression and treat-
ment response (Yu et al., 2020). Additionally, in neu-
rodegenerative diseases such as Alzheimer’s, RNA-
seq has revealed critical interactions between gene
pairs that contribute to disease mechanisms, offering
potential targets for intervention (Chen et al., 2019).
Furthermore, in cardiovascular research, RNA-seq
has provided insights into genes associated with heart
failure and atrial fibrillation, aiding in the develop-
ment of predictive models that enhance disease pre-
diction and support precision medicine (Venkat et al.,
2023).
Building on these advancements, ML models play
a crucial role in leveraging RNA-seq data for dis-
ease research. Supervised learning models, such as
SVMs and RFs, have been effectively utilized to
identify biomarkers and predict treatment responses.
Gupta et al. (Gupta et al., 2021) deployed RF and
SVM to analyze RNA-seq datasets for identifying
and validating novel transcript biomarkers associated
with hepatocellular carcinoma, focusing on improv-
ing early detection and diagnosis. Meanwhile, un-
supervised learning models like hierarchical cluster-
ing have uncovered novel gene expression patterns in
cancer research, offering valuable insights into under-
lying mechanisms. Lee et al. (Lee et al., 2020) pro-
posed an approach that uses similarity-based hierar-
chical clustering to accurately analyze complex pa-
tient data and identify distinct patterns that correlate
with disease progression, thereby enhancing the pre-
diction of pathological stages in papillary renal cell
carcinoma.
To enhance the efficacy of ML models applied
to RNA-seq datasets, it is essential to conduct sev-
eral preprocessing steps on the raw data. These steps
typically include quality control measures to remove
low-quality reads, serving as an initial data denoising
process. In addition, one can deploy feature normal-
ization to account for variations in sequencing depth
and filtering to eliminate uninformative genes. More-
over, feature extraction methods, such as gene expres-
sion quantification and dimensionality reduction tech-
Machine Learning-Based Prediction of Key Genes Correlated to the Subretinal Lesion Severity in a Mouse Model of Age-Related Macular
Degeneration
629

niques such as principal component analysis (PCA)
(Chen et al., 2020), are crucial for identifying relevant
features that capture the most informative aspects of
the RNA-seq data.
2.3 Molecular Studies on the
Pathogenesis of Subretinal Fibrosis
Subretinal fibrosis is a hallmark of advanced neo-
vascular age-related macular degeneration (nAMD)
and is closely associated with poor visual outcomes
(Tenbrock et al., 2022). Although anti-VEGF thera-
pies have revolutionized the treatment of nAMD by
suppressing neovascularization, their long-term ef-
fectiveness is limited. A substantial proportion of
patients, despite initial responsiveness to anti-VEGF
therapies, develop subretinal fibrosis within a decade,
leading to irreversible vision loss (Khachigian et al.,
2023). Fibrosis, characterized by the excessive accu-
mulation of extracellular matrix (ECM) components
beneath the retina, results in tissue scarring and visual
impairment (Mallone et al., 2021). This underscores
the need for a deeper understanding of the molecular
pathways contributing to fibrotic processes, as current
treatment strategies are insufficient in halting or re-
versing fibrosis progression.
Recent research has uncovered critical molecular
factors that influence the severity of subretinal fibro-
sis. Specifically, mutations in genes responsible for
ECM remodeling have been implicated as key drivers
of fibrosis in nAMD (Shughoury et al., 2022). Col-
lagen and fibronectin, structural proteins of the ECM,
are essential for maintaining tissue integrity and fa-
cilitating repair processes (Nita et al., 2014). How-
ever, genetic mutations in these proteins can promote
aberrant ECM deposition, exacerbating fibrotic tissue
development. Furthermore, the interplay between ge-
netic predispositions and environmental factors, such
as oxidative stress and chronic inflammation, accel-
erates the fibrotic response in the subretinal space
(Kauppinen et al., 2016). Inflammatory signaling
pathways, such as those mediated by cytokines and
chemokines, are known to amplify the fibrotic pro-
cess by enhancing the recruitment of fibroblasts and
the deposition of ECM components, leading to reti-
nal scarring. This interaction between genetic and en-
vironmental factors highlights the complexity of fi-
brosis and the need for multifaceted therapeutic ap-
proaches.
3 METHODOLOGY
3.1 Framework Overview
Our study, as depicted in Figure 1, employs a compre-
hensive ML-based framework designed to predict le-
sion severity and identify key gene targets associated
with the disease using RNA-seq data from the JR5558
mouse model. This approach aims to deepen our un-
derstanding of the genetic factors influencing disease
progression and to uncover potential therapeutic tar-
gets.
In the initial phase of our research, we collected
RNA-seq data from the retinas of 23 JR5558 mice.
This data was meticulously correlated with lesion
severity scores obtained from fundus photographs of
these mice, where severity was quantified by mea-
suring subretinal lesion size. This process provides
us with a foundational raw RNA-seq dataset, crucial
for subsequent analyses. Next, in the feature engi-
neering stage, we tackled the challenge posed by the
limited sample size and the extensive number of fea-
tures. This was achieved through dimensionality re-
duction, specifically by organising the data accord-
ing to group genes into different molecular pathways.
This method allows us to focus on the most perti-
nent gene groups. Once these influential gene groups
were identified, we expanded the dataset to concen-
trate on individual genes within these groups, enhanc-
ing the dataset’s utility for further analysis. Following
this, taking the refined dataset as input, we proceeded
to the training and prediction phase using two ML
models: Ridge regression and ElasticNet regression.
These models were chosen for their ability to handle
complex data and provide accurate predictions. Then,
based on our trained models, we conduct two itera-
tive experiments: biological correlation and influence
measurement. The objective of the first experiment is
to identify genes whose expression is most strongly
associated with the severity of subretinal lesions in
AMD, while the second experiment aims to identify
target genes that, when modified, could significantly
alter lesion severity, potentially revealing new treat-
ment targets for subretinal fibrosis in AMD. Finally,
we delved into a comprehensive discussion of the bio-
logical impact of our findings. This analysis is pivotal
in exploring and identifying potential therapeutic tar-
gets, which may offer new avenues for the treatment
of subretinal fibrosis.
By integrating these stages, our framework pro-
vides a robust and systematic approach to understand-
ing the genetic influences on lesion severity, ulti-
mately contributing valuable insights for future thera-
peutic interventions.
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
630
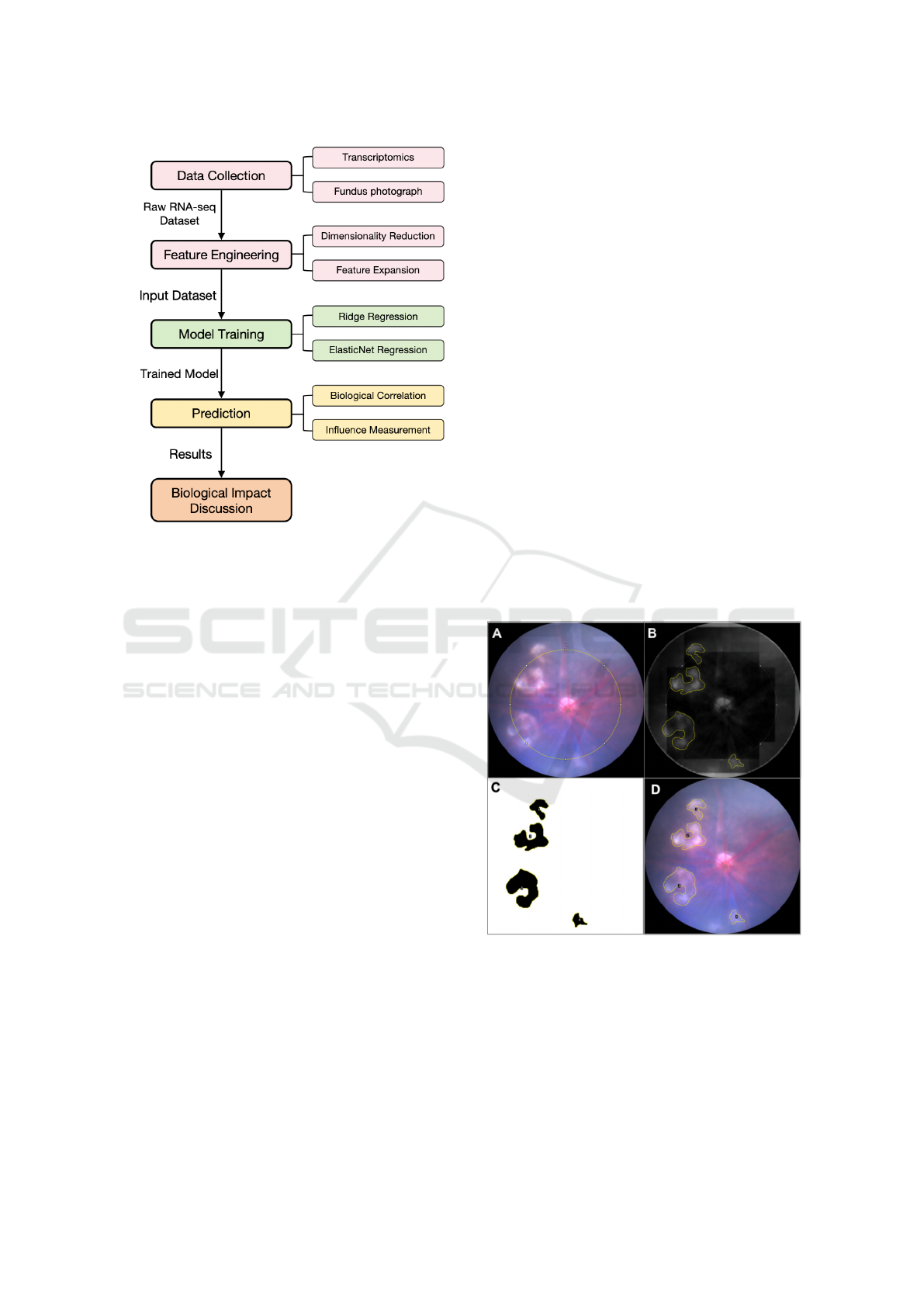
Figure 1: The overall framework of our study.
3.2 Animals
JR5558 mice were purchased from Jackson Lab-
oratory (B6.Cg-Crb1
rd8
Jak3
m1J
/Boc: JAX stock#
005558), with a genetic background of C57BL/6J.
All animals were housed in the pathogen-free envi-
ronment of the Animal Research Facility on a 12-
light/dark cycle. The experimental procedures were
conducted in accordance with the ARVO Statement
for the Use of Animals in Ophthalmic and Vision
Research (http://www.arvo.org/). This study was ap-
proved by the Animal Ethics Committee of the Uni-
versity of Sydney (Project number: 2021/2013).
3.3 Data Collection
The study utilizes bulk RNA-seq data from the
retinas of JR5888 mice, a well-established model
for studying nAMD. Total RNA from twenty-three
retinas of 8-week-old male JR5558 mice was ex-
tracted using GenEluteTM Single Cell RNA Purifi-
cation Kit (Sigma Aldrich, RNB300). The library
preparation, quality control and sequencing were
commercially contracted to Novogene (https://www.
novogene.com/). The RNA-seq dataset includes ex-
pression levels of 56,748 genes, quantified as Frag-
ments Per Kilobase of transcript per Million mapped
reads (FPKM). The FPKM values provide a normal-
ized measure of gene expression, accounting for gene
length and sequencing depth, allowing for accurate
comparisons across samples. Out of the 56,748 genes,
24,888 have corresponding Entrez IDs, which were
used for subsequent pathway analysis. In addition
to the RNA-seq data, fundus photographs were also
taken to analyze the disease severity. In brief, mice
were anesthetised by intraperitoneal injection of ke-
tamine (48 mg/kg, Troy Laboratories, Australia) and
medetomidine (0.6 mg/kg, Troy Laboratories, Aus-
tralia). Mice pupils were dilated with 0.5% Tropi-
camide. Fundus photographs were performed with
MICRON IV Retinal Imaging Microscope (Phoenix
Technology Group, USA) with optic nerve locating at
the center of the image. The total area of subretinal le-
sions on fundus images was then independently quan-
tified by two investigators using FIJI ImageJ software
( https://imagej.net/software/fiji/downloads, National
Institutes of Health, USA). Figure 2 illustrates the key
steps of the quantification method. An imageJ macro
was developed accordingly to ensure consistent out-
put. Of note, lesions that either totally or partially fell
into the circle with a radius of 283 µm from the op-
tic nerve were included in the analysis. Lesion sever-
ity was quantified based on the percentage of subreti-
nal lesion area observed in fundus photographs of the
mouse retina. The lesion area serves as the primary
outcome variable for predicting the severity of fibro-
sis in this study.
A: Draw a circle with a radius of 283 µm centered on the
optic nerve. B: Convert the image to an 8-bit grayscale,
remove the background and mark with freehand tools. C:
Adjust the threshold to precisely select the lesion area. D:
Verify in the original image that all lesions have been se-
lected.
Figure 2: Key steps of the quantification method.
Machine Learning-Based Prediction of Key Genes Correlated to the Subretinal Lesion Severity in a Mouse Model of Age-Related Macular
Degeneration
631

3.4 Feature Engineering
3.4.1 Dimensionality Reduction
Dimensionality reduction is a crucial technique in the
analysis of RNA-seq data, particularly when investi-
gating the influence of genes on disease severity. In
relation to our research on the JR5558 mouse model,
dimensionality reduction serves to strike a balance
between model performance and computational effi-
ciency. High-dimensional data can lead to overfitting,
where the model becomes too tailored to the training
data of small size, resulting in poor generalisation to
unseen data. Conversely, reducing the dimensional-
ity excessively may lead to the loss of vital biological
information necessary for understanding disease pro-
gression.
Several methods are available for dimensionality
reduction, including feature extraction techniques like
PCA and feature selection methods such as Least Ab-
solute Shrinkage and Selection Operator (LASSO)
and RF. For our study, feature selection is particularly
advantageous, as it retains the interpretability of the
model. This is essential for identifying potential ther-
apeutic targets, as we aim to elucidate the relationship
between individual genes and the severity of subreti-
nal fibrosis.
Incorporating domain knowledge is essential for
effective feature selection from RNA-seq data, as it
helps identify relevant genes that influence disease
severity. Without this understanding, ML models may
miss important non-linear relationships, potentially
excluding critical genes. To maintain robust model
performance, feature selection should be performed
solely on the training dataset to prevent data leakage,
which could skew performance metrics. Additionally,
ensuring consistent variable types across training and
testing datasets is crucial for achieving reliable pre-
dictions and generalizability.
In our study, we face a challenge common to many
biological datasets: a limited number of samples
from JR5558 mice, yet a vast array of gene features
for each sample. To address this, we developed an
approach to effectively reduce the dimensionality of
the RNA-seq data by grouping genes into canonical
molecular pathways. A web scraper was developed in
R to extract detailed biological pathway information
related to Mus musculus from the KEGG database
(https://www.genome.jp/kegg-bin/show organism?
menu type=pathway maps&org=mmu). The data
extracted included pathway maps, which were orga-
nized into a data frame for subsequent dimensional
reduction. This step was crucial to managing the high
dimensionality of the RNA-seq data by focusing on
relevant pathways rather than individual genes, thus
reducing noise and improving the model’s ability
to identify significant correlations. By grouping
gene features and averaging expression values within
each group, we were able to reduce the dataset’s
dimensionality from over 24,888 features to 343,
thereby streamlining the dataset for more effective
prediction and analysis.
3.4.2 Feature Expansion
Feature expansion is a technique used to enhance the
predictive capabilities of a model by increasing the
number of relevant features. It involves transform-
ing existing data to uncover additional insights that
may not be immediately apparent. This method can
be particularly useful in complex biological datasets,
allowing for a more comprehensive analysis by incor-
porating diverse aspects of the data.
Pathway-based dimensionality reduction allows
for effective predictive analysis on gene-group
datasets with reduced dimensionality. Once we iden-
tify the key gene groups, we can expand their original
gene features to enhance training and prediction fo-
cused on these crucial genes. Specifically, genes from
the identified key biological pathways are extracted
and their FPKM values from each sample are used as
features for the second round of data processing. This
gene-based feature expansion allows the model to in-
corporate detailed expression information for genes
that are potentially involved in fibrosis, enhancing its
predictive power.
4 EXPERIMENTS
4.1 Dataset Description
The dataset for this study comprises bulk RNA-seq
data from the retinas of JR5888 mice and correspond-
ing lesion area measurements. The RNA-seq data
included expression levels for 56,748 genes, with
24,888 genes associated with Entrez IDs. The le-
sion area data were derived from fundus photographs,
where the extent of subretinal fibrosis was quantified
as a percentage of the total retinal area. This dataset
was divided into training and validation sets to eval-
uate the performance of the ML models in predicting
key genes associated with disease severity.
4.2 Prediction and Results
In this study, we conducted two prediction tasks: 1.
biological significance and 2. influence measurement,
each involving two rounds of iterative experiments.
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
632
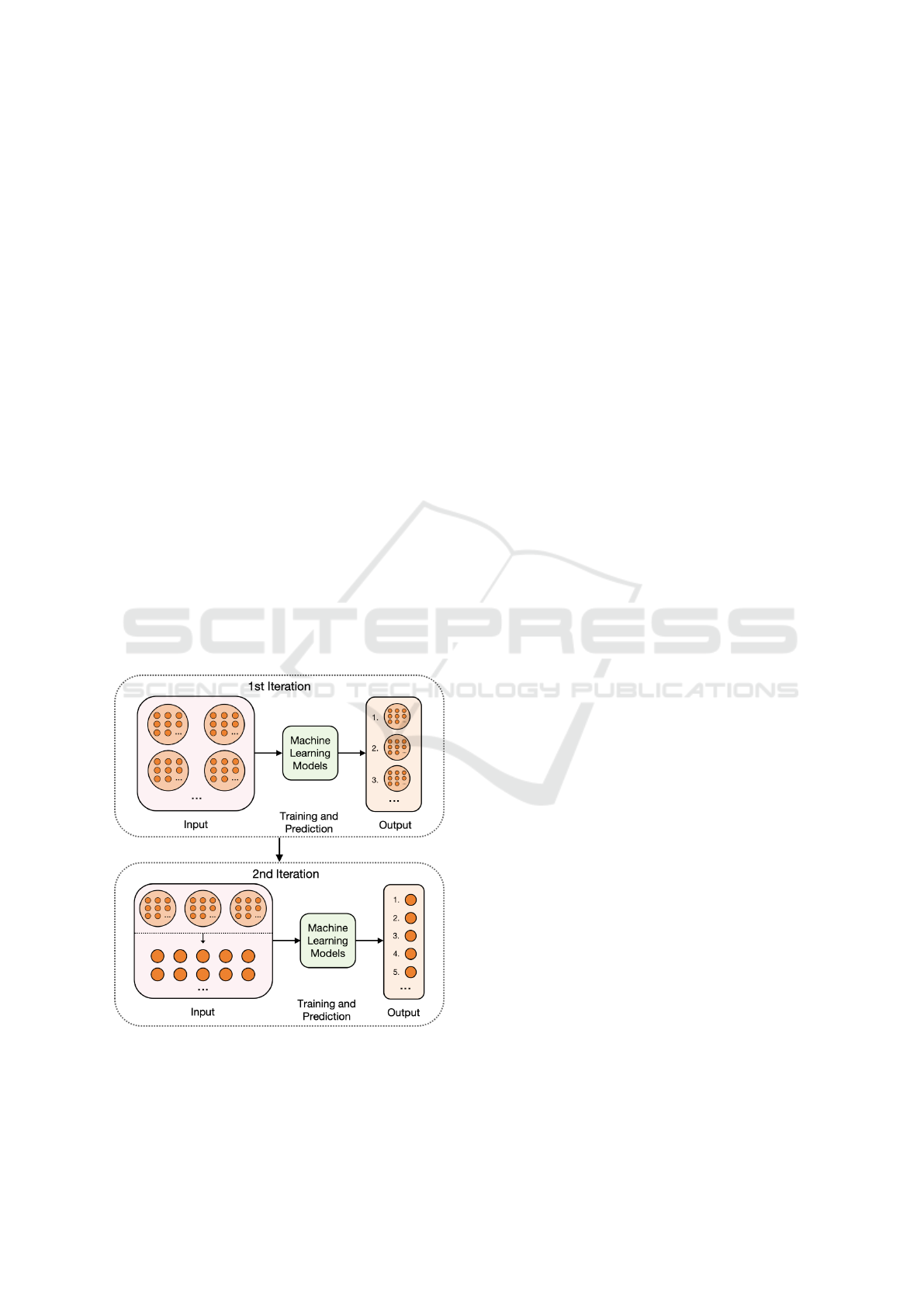
Fig. 3 illustrates the logic of our experimental ap-
proach. In the first round, we employed a dataset
with gene groups as features, utilizing dimensional-
ity reduction through a gene pathway-based method
to perform predictive tasks. Based on these initial re-
sults, we identified the top-performing gene groups as
candidates for further analysis. In the second round,
we expanded our focus by using the expression values
of the original genes within these selected candidates.
This allowed us to conduct more detailed prediction
tasks and perform biological analysis targeting indi-
vidual genes, thereby enhancing the depth and accu-
racy of our findings.
In this study, we conducted two prediction tasks:
1. biological significance and 2. influence measure-
ment, each involving two rounds of iterative experi-
ments. Figure 3 illustrates the logic of our experimen-
tal approach. In the first round, we employed a dataset
with gene groups as features, utilizing dimensional-
ity reduction through a gene pathway-based method
to perform predictive tasks. Based on these initial re-
sults, we identified the top-performing gene groups as
candidates for further analysis. In the second round,
we expanded our focus by using the expression values
of the original genes within these selected candidates.
This allows us to conduct more detailed prediction
tasks and perform biological analysis targeting indi-
vidual genes, thereby enhancing the depth and accu-
racy of our findings.
Figure 3: The logic of our iterative experiments.
4.2.1 Biological Correlation
The objective of the first task is to predict the genes
most strongly correlated with the severity of disease
progression in AMD.
In the first round of experiments, we used Ridge
regression and ElasticNet regression models to train
and predict the gene group dataset following dimen-
sionality reduction, identifying the overlapping most
influential gene groups from both models. We iden-
tified nine recurring candidates from the top ten most
influential gene groups produced by both models. In
the second round of experiments, we expanded the
gene expression features within these influential gene
groups to further identify the most significant genes.
The seven selected most influential genes, which have
the greatest association with the disease in biological
correlation experiments, are listed in Table 1.
4.2.2 Influence Measurement
The purpose of the second task is to identify tar-
get genes that, when manipulated, could significantly
modulate the severity of subretinal fibrosis, poten-
tially leading to more effective therapeutic strategies.
In the first round, we adjusted each gene group
feature by decreasing its expression value to 50% and
increasing it to 200%, respectively. We then applied
the trained model to predict lesion severity based on
these modified gene group expressions and recorded
the changes in predicted severity. We calculated the
differences in lesion severity before and after the ad-
justments to each gene group’s expression. This al-
lows us to identify the gene groups whose modifica-
tions resulted in the most significant changes in lesion
severity. Here, we counted and ranked the changes in
lesion severity into two categories: aggravation and
alleviation. Therefore, we identified the most signif-
icant gene groups that either worsened or alleviated
the disease as candidates. By the end of the first
round of experiments, we obtained two rankings for
the gene groups: 50% manipulation that alleviated the
disease, and 200% manipulation that caused the dis-
ease to worsen.
In the second round of experiments, we performed
feature expansion on the two ranked gene group lists
from the first round and obtained two correspond-
ing datasets with individual gene expression values as
features. For each dataset, we performed the same
manipulations as in the previous round and counted
the rankings that caused the same disease impact.
This time, we identified the most influential individ-
ual genes. For example, for the gene groups that re-
sulted in 50% manipulation causing the disease alle-
viation in the previous round, we continued to apply
Machine Learning-Based Prediction of Key Genes Correlated to the Subretinal Lesion Severity in a Mouse Model of Age-Related Macular
Degeneration
633

Table 1: Top 7 most important genes selected by our ML models in biological correlation experiments with the greatest
association to the disease.
Ranking Influential Gene Entrez ID Gene Name and Description
1 16177 interleukin 1 receptor, type I
2 18796 phospholipase C, beta 2
3 12259 complement component 1, q subcomponent, alpha polypeptide
4 320302 glycosyltransferase 28 domain containing 2
5 15212 hexosaminidase B
6 18018 nuclear factor of activated T cells, cytoplasmic, calcineurin dependent 1
7 20848 signal transducer and activator of transcription 3
Table 2: Top 10 genes selected by our ML models for their significant impact on disease improvement when gene expression
values were reduced by 50%.
Ranking Influential Gene Entrez ID Gene Name and Description
1 66357 oligosaccharyltransferase complex subunit (non-catalytic)
2 26416 mitogen-activated protein kinase 14
3 12260 complement component 1, q subcomponent, beta polypeptide
4 16179 interleukin-1 receptor-associated kinase 1
5 320302 glycosyltransferase 28 domain containing 2
6 12262 complement component 1, q subcomponent, C chain
7 224530 acetyl-Coenzyme A acetyltransferase 3
8 14676 guanine nucleotide binding protein, alpha 15
9 16176 interleukin 1 beta
10 12503 CD247 antigen
Table 3: Top 10 genes selected by our ML models for their significant impact on disease exacerbation when gene expression
values were increased to 200%.
Ranking Influential Gene Entrez ID Gene Name and Description
1 18798 phospholipase C, beta 4
2 12260 complement component 1, q subcomponent, beta polypeptide
3 12259 complement component 1, q subcomponent, alpha polypeptide
4 12322 calcium/calmodulin-dependent protein kinase II alpha
5 12262 complement component 1, q subcomponent, C chain
6 19091 protein kinase, cGMP-dependent, type I
7 106759 toll-like receptor adaptor molecule 1
8 20293 chemokine (C-C motif) ligand 12
9 224530 acetyl-Coenzyme A acetyltransferase 3
10 12789 cyclic nucleotide gated channel alpha 2
the same 50% manipulation after expanding the gene
dataset and ranked the genes that had the same impact
on the disease, which was improving. This process al-
lowed us to identify the genes that most significantly
affected lesion severity. Tables 2 and 3 have shown
the final results of the influence measurement experi-
ments, respectively.
4.3 Biological Impact Discussion
The integration of ML models with RNA-seq data of-
fers a robust approach for identifying transcriptomic
factors linked to disease severity in AMD. This study
specifically aimed at predicting genes associated with
the severity of subretinal lesions in a mouse model
of AMD. By employing dimensionality reduction and
feature expansion techniques, our model successfully
identified genes within specific pathways that their re-
lated proteins and signaling pathways are likely con-
tributors to subretinal fibrosis that may be considered
as therapeutic targets. These findings enhance our un-
derstanding of the molecular mechanisms underlying
subretinal fibrosis and present new opportunities for
therapeutic interventions.
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
634

It is remarkable that two different tasks with
different algorithms identified several common key
genes/proteins or targeting same pathway. We first
compared the top 10 target genes in table II and table
III and identified two proteins is positively correlated
to the lesion severity. They are Complement compo-
nent 1, q subcomponent (C1q) and Acetyl-Coenzyme
A acetyltransferase 3 (ACAT3). We further compared
the results from task 1 and task 2 and further two
more common proteins has been identified. They are
Phospholipase C (PLC), and Glycosyltransferase 28.
All four common proteins were strongly implicated in
both the progression of subretinal fibrosis and as po-
tential therapeutic targets (Tables 1, 2 and 3). Each
of these proteins operates within distinct yet inter-
connected biological processes, highlighting a com-
plex interaction between inflammation, immune re-
sponses, cellular signaling and metabolic regulation
that drives fibrotic changes in the retina.
The C1q proteins are key components of the clas-
sical complement pathway which initiates immune
surveillance, inflammation and the clearance of apop-
totic cells (Cho, 2019) (Galvan et al., 2012). Dys-
regulation of this pathway, particularly sustained ac-
tivation of C1q, has been closely linked to the pro-
gression of AMD (Ma et al., 2022). Ongoing C1q
activation in subretinal fibrosis fuels chronic inflam-
mation, promoting tissue damage and extracellular
matrix remodeling—both hallmarks of fibrosis. The
accumulation of matrix components in the subretinal
space results in tissue thickening and scarring, which
contribute to irreversible loss of vision in advanced
AMD. It is worth noting that we also identified sev-
eral inflammation-related molecules, including Inter-
leukin 1 receptor, type I (IL1R1, Table 1), Signal
transducer and activator of transcription 3 (STAT3,
Table 1), Interleukin-1 receptor-associated kinase 1
(IRAK1, Table 2), Interleukin 1 beta (IL1B, Table 2),
Toll-like receptor adaptor molecule 1 (TICAM1, Ta-
ble 3) and Chemokine (C-C motif) ligand 12 (CCL12,
Table 3). This suggests a strong positive correlation
between the activation of inflammatory pathways and
the severity of the subretinal lesion.
PLC is critical for intracellular signal transduc-
tion. This signaling pathway is also essential for regu-
lating cellular responses to inflammatory stimuli, pro-
liferation and apoptosis (Wu et al., 2023). PLC may
intensify complement activation in subretinal fibrosis
by amplifying pro-inflammatory signals (Zhu et al.,
2018). This interaction between PLC and the com-
plement system may create a self-perpetuating cycle
of inflammation that drives tissue remodeling and fi-
brosis in the retina.
Acetyl-Coenzyme A acetyltransferase (ACAT)
plays a key role in acetyl-CoA metabolism by in-
fluencing cholesterol and production of ketone bod-
ies (Ma et al., 2023). Its involvement in lipid
metabolism is particularly relevant to retinal diseases,
where metabolic dysregulation often exacerbates in-
flammation and tissue damage (Ana et al., 2023). Al-
tered ACAT activity could disrupt lipid homeostasis,
promoting cellular stress and inflammation in retinal
cells, which may accelerate the development of sub-
retinal fibrosis by further stimulating inflammatory
and fibrotic processes.
Glycosyltransferase is an enzyme responsible for
glycosylation, the addition of sugar moieties to pro-
teins and lipids. This modification impacts pro-
tein folding, stability and interactions, all of which
are crucial for maintaining cellular function (Nagare
et al., 2021). Aberrant glycosylation has been asso-
ciated with fibrosis across various tissues (Loaeza-
Reyes et al., 2021). In subretinal fibrosis, Glycosyl-
transferase may influence key protein modifications
involved in inflammation and extracellular matrix for-
mation, potentially exacerbating tissue scarring and
the progression of fibrosis.
Together, these four proteins, C1q, PLC, ACAT3
and Glycosyltransferase, likely form an intercon-
nected network that sustains chronic inflammation
and metabolic dysfunction in the retina. Their com-
bined activity promotes extracellular matrix deposi-
tion and tissue remodeling, driving the progression of
subretinal fibrosis. Understanding their precise roles
and interactions could provide critical insights into
therapeutic strategies for AMD. Targeting these pro-
teins or their signaling pathways may offer effective
ways to reduce inflammation, slow fibrotic changes
and prevent vision loss in patients with AMD.
These results underscore the potential of combin-
ing ML with molecular imaging techniques to en-
hance our understanding of fibrotic diseases and im-
prove patient outcomes. Future research could ex-
plore the application of this approach to other fibrotic
conditions and assess its potential for personalising
treatment strategies based on individual genetic pro-
files.
5 CONCLUSIONS
In this paper, we have presented a comprehensive ap-
proach to addressing the challenges of predicting le-
sion severity in nAMD using an ML-based frame-
work. We introduced a unique RNA-seq dataset de-
rived from the JR5558 mouse model, which provides
a valuable resource for further research in subretinal
fibrosis. We successfully addressed issues of limited
Machine Learning-Based Prediction of Key Genes Correlated to the Subretinal Lesion Severity in a Mouse Model of Age-Related Macular
Degeneration
635

sample sizes and high dimensionality typical of RNA-
seq data by employing dimensionality reduction and
feature expansion techniques. Utilizing Ridge and
ElasticNet regression models, our iterative experi-
ments confirmed the effectiveness of our framework,
highlighting its potential in identifying critical genetic
targets linked to subretinal fibrosis.
The insights gained from our study have substan-
tial implications for genetic research and therapeutic
development. We offer new avenues for drug discov-
ery and improved treatment strategies for nAMD by
pinpointing key gene targets, ultimately aiming to en-
hance patient care. Our research underscores the im-
portance of integrating advanced ML techniques in
genomic studies, paving the way for future investiga-
tions that further connect genetic findings with clini-
cal applications.
REFERENCES
Ana, R. d., Gliszczy
´
nska, A., Sanchez-Lopez, E., Gar-
cia, M. L., Krambeck, K., Kovacevic, A., and Souto,
E. B. (2023). Precision Medicines for Retinal Lipid
Metabolism-Related Pathologies. Journal of Person-
alized Medicine, 13(4):635.
Andrews, T. S., Kiselev, V. Y., McCarthy, D., and Hemberg,
M. (2021). Tutorial: guidelines for the computational
analysis of single-cell RNA sequencing data. Nature
Protocols, 16(1):1–9.
Arumugam, K., Naved, M., Shinde, P. P., Leiva-Chauca, O.,
Huaman-Osorio, A., and Gonzales-Yanac, T. (2023).
Multiple disease prediction using machine learning al-
gorithms. Materials Today: Proceedings, 80:3682–
3685.
Bhatt, C. M., Patel, P., Ghetia, T., and Mazzeo, P. L.
(2023). Effective heart disease prediction using ma-
chine learning techniques. Algorithms, 16(2):88.
Blindness, G. (2021). Vision Impairment C, Vision Loss
Expert Group of the Global Burden of Disease S.
Causes of blindness and vision impairment in 2020
and trends over 30 years, and prevalence of avoidable
blindness in relation to VISION 2020: the Right to
Sight: an analysis for the Global Burden of Disease
Study. Lancet Glob Health, 9(2):e144–e160.
Bloch, S. B., Lund-Andersen, H., Sander, B., and Larsen,
M. (2013). Subfoveal fibrosis in eyes with neovascu-
lar age-related macular degeneration treated with in-
travitreal ranibizumab. American Journal of Ophthal-
mology, 156(1):116–124.
Bostanci, E., Kocak, E., Unal, M., Guzel, M. S., Acici, K.,
and Asuroglu, T. (2023). Machine learning analysis
of RNA-seq data for diagnostic and prognostic pre-
diction of colon cancer. Sensors, 23(6):3080.
Chandrasekhar, N. and Peddakrishna, S. (2023). Enhanc-
ing heart disease prediction accuracy through ma-
chine learning techniques and optimization. Pro-
cesses, 11(4):1210.
Chen, H., He, Y., Ji, J., and Shi, Y. (2019). A machine
learning method for identifying critical interactions
between gene pairs in Alzheimer’s disease prediction.
Frontiers in Neurology, 10:1162.
Chen, X., Zhang, B., Wang, T., Bonni, A., and Zhao, G.
(2020). Robust principal component analysis for accu-
rate outlier sample detection in RNA-Seq data. BMC
Bioinformatics, 21(1):269.
Cho, K. (2019). Emerging roles of complement protein C1q
in neurodegeneration. Aging and Disease, 10(3):652–
663.
Galvan, M. D., Greenlee-Wacker, M. C., and Bohlson, S. S.
(2012). C1q and phagocytosis: the perfect comple-
ment to a good meal. Journal of Leukocyte Biology,
92(3):489–497.
Gillies, M., Arnold, J., Bhandari, S., Essex, R. W., Young,
S., Squirrell, D., Nguyen, V., and Barthelmes, D.
(2020). Ten-year treatment outcomes of neovascular
age-related macular degeneration from two regions.
American Journal of Ophthalmology, 210:116–124.
Gupta, R., Kleinjans, J., and Caiment, F. (2021). Identify-
ing novel transcript biomarkers for hepatocellular car-
cinoma (HCC) using RNA-Seq datasets and machine
learning. BMC Cancer, 21(1):962.
Hasegawa, E., Sweigard, H., Husain, D., Olivares, A. M.,
Chang, B., Smith, K. E., Birsner, A. E., D’Amato,
R. J., Michaud, N. A., Han, Y., Vavvas, D. G., Miller,
J. W., Haider, N. B., and Connor, K. M. (2014).
Characterization of a spontaneous retinal neovascular
mouse model. PLoS One, 9(9):e106507.
Islam, M. A., Majumder, M. Z. H., and Hussein, M. A.
(2023). Chronic kidney disease prediction based on
machine learning algorithms. Journal of Pathology
Informatics, 14:100189.
Kauppinen, A., Paterno, J. J., Blasiak, J., Salminen, A.,
and Kaarniranta, K. (2016). Inflammation and its role
in age-related macular degeneration. Cellular and
Molecular Life Sciences, 73:1765–1786.
Khachigian, L. M., Liew, G., Teo, K. Y., Wong, T. Y.,
and Mitchell, P. (2023). Emerging therapeutic strate-
gies for unmet need in neovascular age-related macu-
lar degeneration. Journal of Translational Medicine,
21(1):133.
Khan, A., Qureshi, M., Daniyal, M., and Tawiah, K. (2023).
A novel study on machine learning algorithm-based
cardiovascular disease prediction. Health & Social
Care in the Community, 2023(1):1406060.
Lee, S., Jung, J., Park, I., Park, K., and Kim, D.-S. (2020). A
deep learning and similarity-based hierarchical clus-
tering approach for pathological stage prediction of
papillary renal cell carcinoma. Computational and
Structural Biotechnology Journal, 18:2639–2646.
Linder, M., Bennink, L., Foxton, R. H., Kirkness, M., and
Westenskow, P. D. (2024). In vivo monitoring of
active subretinal fibrosis in mice using collagen hy-
bridizing peptides. Lab Animal, 53(8):196–204.
Loaeza-Reyes, K. J., Zenteno, E., Moreno-Rodr
´
ıguez,
A., Torres-Rosas, R., Argueta-Figueroa, L., Salinas-
Mar
´
ın, R., Castillo-Real, L. M., Pina-Canseco, S., and
Cervera, Y. P. (2021). An overview of glycosylation
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
636

and its impact on cardiovascular health and disease.
Frontiers in Molecular Biosciences, 8:751637.
Ma, Y., Ding, X., Shao, M., Qiu, Y., Li, S., Cao, W., and
Xu, G. (2022). Association of serum complement C1q
and C3 level with age-related macular degeneration
in women. Journal of Inflammation Research, pages
285–294.
Ma, Z., Huang, Z., Zhang, C., Liu, X., Zhang, J., Shu, H.,
Ma, Y., Liu, Z., Feng, Y., Chen, X., et al. (2023). Hep-
atic Acat2 overexpression promotes systemic choles-
terol metabolism and adipose lipid metabolism in
mice. Diabetologia, 66(2):390–405.
Mallone, F., Costi, R., Marenco, M., Plateroti, R., Minni,
A., Attanasio, G., Artico, M., and Lambiase, A.
(2021). Understanding drivers of ocular fibrosis: cur-
rent and future therapeutic perspectives. International
Journal of Molecular Sciences, 22(21):11748.
Mi, X., Zou, B., Zou, F., and Hu, J. (2021). Permutation-
based identification of important biomarkers for com-
plex diseases via machine learning models. Nature
Communications, 12(1):3008.
Nagai, N., von Leithner, P. L., Izumi-Nagai, K., Hosking,
B., Chang, B., Hurd, R., Adamson, P., Adamis, A. P.,
Foxton, R. H., Ng, Y. S., and Shima, D. T. (2014).
Spontaneous CNV in a novel mutant mouse is asso-
ciated with Early VEGF-A-driven angiogenesis and
late-stage focal edema, neural cell loss, and dysfunc-
tion. Investigative Ophthalmology & Visual Science,
55(6):3709–3719.
Nagare, M., Ayachit, M., Agnihotri, A., Schwab, W., and
Joshi, R. (2021). Glycosyltransferases: the multi-
faceted enzymatic regulator in insects. Insect Molec-
ular Biology, 30(2):123–137.
Nita, M., Strzałka-Mrozik, B., Grzybowski, A., Mazurek,
U., and Romaniuk, W. (2014). Age-related macu-
lar degeneration and changes in the extracellular ma-
trix. Medical Science Monitor: International Med-
ical Journal of Experimental and Clinical Research,
20:1003.
Pun, F. W., Ozerov, I. V., and Zhavoronkov, A. (2023). AI-
powered therapeutic target discovery. Trends in Phar-
macological Sciences, 44(9):561–572.
Rafique, R., Islam, S. R., and Kazi, J. U. (2021). Ma-
chine learning in the prediction of cancer therapy.
Computational and Structural Biotechnology Journal,
19:4003–4017.
Rossato, F. A., Su, Y., Mackey, A., and Ng, Y.
S. E. (2020). Fibrotic changes and endothelial-to-
mesenchymal transition promoted by VEGFR2 antag-
onism alter the therapeutic effects of VEGFA pathway
blockage in a mouse model of choroidal neovascular-
ization. Cells, 9(9):2057.
Shughoury, A., Sevgi, D. D., and Ciulla, T. A. (2022).
Molecular genetic mechanisms in age-related macular
degeneration. Genes, 13(7):1233.
Slovin, S., Carissimo, A., Panariello, F., Grimaldi, A.,
Bouch
´
e, V., Gambardella, G., and Cacchiarelli, D.
(2021). Single-cell RNA sequencing analysis: a step-
by-step overview. RNA Bioinformatics, pages 343–
365.
Tenbrock, L., Wolf, J., Boneva, S., Schlecht, A., Agostini,
H., Wieghofer, P., Schlunck, G., and Lange, C. (2022).
Subretinal fibrosis in neovascular age-related macular
degeneration: current concepts, therapeutic avenues,
and future perspectives. Cell and Tissue Research,
387(3):361–375.
Venkat, V., Abdelhalim, H., DeGroat, W., Zeeshan, S., and
Ahmed, Z. (2023). Investigating genes associated with
heart failure, atrial fibrillation, and other cardiovas-
cular diseases, and predicting disease using machine
learning techniques for translational research and pre-
cision medicine. Genomics, 115(2):110584.
Won, J., Shi, L. Y., Hicks, W., Wang, J., Hurd, R., Naggert,
J. K., Chang, B., and Nishina, P. M. (2011). Mouse
model resources for vision research. Journal of Oph-
thalmology, 2011(1):391384.
Wu, Y.-N., Su, X., Wang, X.-Q., Liu, N.-N., and Xu, Z.-W.
(2023). The roles of phospholipase C-β related sig-
nals in the proliferation, metastasis and angiogenesis
of malignant tumors, and the corresponding protective
measures. Frontiers in Oncology, 13:1231875.
Yu, Z., Wang, Z., Yu, X., and Zhang, Z. (2020). RNA-
Seq-Based Breast Cancer Subtypes Classification Us-
ing Machine Learning Approaches. Computational
Intelligence and Neuroscience, 2020(1):4737969.
Zhang, X., Jonassen, I., and Goksøyr, A. (2021). Ma-
chine learning approaches for biomarker discovery us-
ing gene expression data. Bioinformatics.
Zhu, L., Jones, C., and Zhang, G. (2018). The role of
phospholipase C signaling in macrophage-mediated
inflammatory response. Journal of Immunology Re-
search, 2018(1):5201759.
Machine Learning-Based Prediction of Key Genes Correlated to the Subretinal Lesion Severity in a Mouse Model of Age-Related Macular
Degeneration
637
