
Assessing the Influence of scRNA-Seq Data Normalization on
Dimensionality Reduction Outcomes
Marcel Ochocki
1a
, Michal Marczyk
1,2 b
and Joanna Zyla
1c
1
Department of Data Science and Engineering, Silesian University of Technology, Akademicka 16, Gliwice, Poland
2
Breast Medical Oncology, Yale Cancer Center, Yale School of Medicine, New Haven, CT, U.S.A.
Keywords: Unsupervised Learning, Data Normalization, Dimensionality Reduction, Single-Cell Sequencing.
Abstract: Through the decades, improvements in high-throughput molecular biology techniques have brought to the
level of sequencing transcripts from single cells (scRNA-Seq) instead of bulk material. Implementing these
new techniques requires innovative analytical methods and knowledge about their performance. Data
normalization is a crucial step in the bioinformatical pipeline applied in scRNA-Seq analysis. We evaluated
the impact of six commonly used normalization methods on two dimensionality reduction methods, namely
tSNE and UMAP, using three real scRNA-Seq datasets. We tested dispersion and clustering efficiency using
three clustering algorithms after dimensionality reduction. Our results demonstrated that simple normalization
methods, such as log2 or Freeman-Tukey, as well as scran normalization consistently outperformed other
scRNA-seq-dedicated techniques, yielding superior dimensionality reduction and clustering efficiency for
small and medium-sized datasets. Regardless of no statistically significant enhancement in results for any
dimensionality reduction methods or clustering techniques, the Louvain clustering method consistently
demonstrated lower performance results. We conclude, that the choice of normalization technique should be
carefully tailored to the dataset’s size and characteristics since it may affect the final within-pipeline
processing results.
1 INTRODUCTION
Recent advances in RNA sequencing technologies
have increased the sensitivity and specificity of
transcriptome analysis. The latest solutions allow for
precise analysis of transcript heterogeneity and reveal
novel subpopulations and cell types on an individual
cell level (single-cell RNA sequencing; scRNA-Seq).
Yet, the introduction of scRNA-Seq brought many
challenges to bioinformatical analysis (Hwang et al.,
2018). One of the first steps in scRNA-Seq analysis
is data normalization which reduces technical noise
and existing biases. Moreover, normalization results
in comparable gene counts within and between cells
that allow for more precise downstream analysis.
Throughout the development of scRNA-seq, a variety
of normalization methods have been employed,
including adaptations of bulk sequencing techniques
(Hafemeister and Satija, 2019) as well as novel
a
https://orcid.org/0009-0001-0814-3431
b
https://orcid.org/0000-0003-2508-5736
c
https://orcid.org/0000-0002-2895-7969
approaches specifically designed for scRNA-Seq
studies. Yet, the first one can overcorrect for scaling
factor sizes (Vallejos et al., 2017). Recently, many
normalization methods were introduced and several
studies tested their efficiency and impact on further
analysis (Cole et al., 2019, Vieth et al., 2019). In
(Lytal et al., 2020) authors assessed using empirical
visualization, impact on classification, and
computational time. In (Brown et al., 2021) authors
introduced a new normalization method (Dino) with
comparison to other solutions and tested their
influence on differential expression analysis based on
a relationship between average TPR and average FPR
for a Wilcoxon rank-sum test, as well as on clustering.
Finally, one of the biggest studies (Ahlmann-Eltze
and Huber, 2023) tested methods for consistency,
simulation, and downsampling.
In the presented manuscript, we concentrated on
the impact of the normalization step on dimensiona-
504
Ochocki, M., Marczyk, M. and Zyla, J.
Assessing the Influence of scRNA-Seq Data Normalization on Dimensionality Reduction Outcomes.
DOI: 10.5220/0013318700003911
Paper published under CC license (CC BY-NC-ND 4.0)
In Proceedings of the 18th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2025) - Volume 1, pages 504-515
ISBN: 978-989-758-731-3; ISSN: 2184-4305
Proceedings Copyright © 2025 by SCITEPRESS – Science and Technology Publications, Lda.

lity reduction outcomes and their clustering ability.
The dimensionality reduction by tSNE or UMAP is
one of the most common ways to present scRNA-Seq
data studies (Cakir et al., 2020). Moreover, it is one
of the most important steps to visualize the
heterogeneity of the analyzed dataset. Thus, joint
solutions were also introduced to compare different
datasets (j-tSNE and j-UMAP) (Do and Canzar,
2021). Yet, to our best knowledge, the impact of
normalization to reductions given by tSNE and
UMAP was not tackled before in non-empirical way.
To reach the gain of the study we collected three
real scRNA-Seq datasets with known cell labels and
different sample sizes, for which we ran six different
normalization techniques. Next, based on normalized
data we extracted the tSNE and UMAP 2D
embeddings and measured the effect of normalization
on dispersion in dimensionality reduction. Moreover,
we checked the impact of normalization on clustering
performance based on 2D transformed data by three
different clustering methods.
2 MATERIALS AND METHODS
2.1 scRNA-Seq Datasets
Three scRNA-Seq datasets of diverse sample sizes
and labeled cell types were used to assess the quality
of performed clustering (Table 1). The first dataset,
called Liver, including immunological cells, was
extracted from liver tissue (Wang et al., 2021) and is
available under access number E-MTAB-10553. The
dataset includes 15,650 labeled cells divided into 13
groups. The second dataset, PBMC, provides
information from peripheral blood mononuclear cells
(Ding et al., 2020). Only experiment 1A performed
on Chromium v2 10x platform, where 3,222 cells
were grouped into 9 cell types, was used here. The
data are available at the single-cell portal of Broad
Institute (https://singlecell.broadinstitute.org). The
smallest dataset includes cells derived from different
tissues of breast cancer (BC) subtypes (HER+,
Luminal A, Luminal B, and Tripple Negative Breast
Cancer) as well as normal ones (Chung et al., 2017).
Due to the presence of samples from healthy tissue,
this dataset was investigated in two ways: (i) with all
possible groups i.e. 5 (BC_sub), and (ii) healthy vs
cancer tissue cells (BC_dis). The dataset is publicly
available under access number E-GEOD-75367.
For every dataset, three pre-processing steps were
performed: (i) transcripts with only zero counts across
all cells and with low variance of normalized
expression were filtered out using GaMRed (Marczyk
et al., 2018); (ii) transcripts without annotation were
removed; (iii) for the transcripts with duplicated
Ensembl ID the one with higher variance were kept.
Table 1: Summary of used scRNA-Seq datasets.
Dataset # of
samples
# of
features
# of cell
types /
classes
Liver 15000 15,650 13
PBMC 3,222 15,817 9
Breast Cancer
(BC_sub)
244 16,639 5
Breast Cancer
(BC_dis)
244 16,639 2
2.2 Data Normalization Methods
Several normalization techniques widely used in
scRNA-seq data analysis were tested (Table 2). Both,
primary methods like the log2 transformation and the
Freeman-Tukey square root (FT) transformation, as
well as several novel normalization techniques
specifically suited for scRNA-seq data, were
included. Before basic transformations, data were
scaled by the median counts across all cells to
mitigate the sequencing-depth normalization and
stabilize the variance across the different gene
expression levels.
2.2.1 Simple Transformations
Logarithmic normalization, particularly the log2
transformation, is a popular choice for reducing
distribution skewness and is typically used in
standard RNA-seq preprocessing pipelines before
downstream feature selection (Luecken et al., 2019,
Lytal et al., 2020, Cuevas-Diaz et al., 2024).
Importantly, before applying the log2 transformation,
a small ‘pseudocount’ of one was added to all gene
counts to account for both technical and cell-specific
absences of transcript counts. This step is a well-
established standard in such a pipeline (Lytal et al.,
2020).
Square root transformation, though less common
than logarithmic one, is another effective
normalization technique in scRNA-seq data
processing (Lause et al., 2021, Booeshaghi et al.,
2022). The choice of square root transformation,
especially the FT transformation, is often vastly
justified by the characteristics of scRNA-seq data,
which are frequently modeled using a Poisson
distribution (Brown et al., 2021, Lause et al., 2021,
Choudhary and Satija, 2022).
Assessing the Influence of scRNA-Seq Data Normalization on Dimensionality Reduction Outcomes
505

2.2.2 Scran – Normalization via
Deconvolution Across Pooled Cells
The Scran normalization method (Lun et al., 2016)
aims to enhance overall normalization efficiency
through a deconvolution process. The core objective
of this approach is to estimate the adjusted cell
transcript count based on cell-specific parameters,
which describe the cell bias and its corresponding
adjustment factor, respectively. However, to obtain
unbiased estimates of true expressions, several
assumptions and computations must be taken into
account. First of all, cell pools are created by
grouping cells with similar library sizes. This is a
pivotal step in Scran normalization, that helps to
reduce variability arising from technical differences,
e.g. sequencing depth. Next, the pool-based size
factor can be determined as the ratio of the sum of
transcript counts within the k-th pool, and
the mean of
gene counts across the entire cell population. The
estimates of the factor within all cell pools are then
calculated as the median across genes, based on the
assumption that the majority of genes are non-
differentially expressed. Based on that, we can
construct a system of linear equations to estimate the
cell-specific biases, that finally can be solved with a
standard least-squares method.
2.2.3 SCnorm – Normalization Using
Quantile Regression with Gene
Grouping
The SCnorm approach (Bacher et al., 2017) models
the relationship between gene log-transformed
expression counts and the corresponding cells' log-
transformed sequencing depth (hereafter, the ‘log-
transformed’ participle will be omitted for
simplicity). The genes are initially grouped into K
pools (by default at the first step K=1) to preserve cell
variability. For each of these K groups, the
relationship between gene expression counts and
sequencing depth is modeled using median quantile
regression for each gene and cell. Additionally,
quantile regression is employed to estimate a similar
relationship for the overall expression of all genes.
SCnorm assumes that the median may not always be
the best estimate for the entire set of genes, thus it
considers multiple quantiles, as well as several
degrees of polynomial, to improve accuracy. The
authors propose that the optimal quantiles and
degrees minimize the difference between the count-
depth relationship value across predicted expressions,
estimated via median regression using a first-degree
polynomial, and the mode of such a relationship for
un-normalized counts. The scale factor for each cell
is computed based on the estimated quantiles for each
group. Specifically, for each gene group, the scale
factor for a cell is defined as the ratio between the
gene expression values at a selected quantile and the
corresponding predicted values from the regression
model. Moreover, to adjust the number of K, a
specific condition is defined; the modes of the slopes
within equal-sized gene groups must be less than 0.1.
If at least one of them is greater, the initial number of
K = 1 is increased by one, and the genes are pooled
across groups with the k-medoids algorithm.
However, the authors suggested considering pre-
defined conditions under which the normalization
procedure may proceed before being applied to the
entire dataset. To maintain the unsupervised nature of
the pipeline, we decided to pre-aggregate cells into
separate groups using hierarchical clustering, as
described in the supplementary materials provided in
the Bioconductor guides.
2.2.4 Dino – Normalization by
Distributional Resampling
Dino (Brown et al., 2021) is an approach that aims to
reconstruct transcript expression distributions that are
independent of the cell’s library size. Those
distributions are Poisson means modeled as Gamma
mixtures. In this study, the number of Gamma
components is set to 100, as a default value proposed
in the original paper. The normalized values of
transcript expression can be sampled from the
posterior distribution with an additional
concentration parameter, that reduces the variability
and centers the normalized values (set to 15, as
originally proposed by the authors).
2.2.5 SCtransform – Normalization with
Variance Stabilization Using
Regularized Negative Binomial
Distribution
The SCtransform normalization method (Hafemeister
and Satija, 2019) utilizes generalized linear models
with regularized Negative Binomial distributions to
model un-normalized transcript counts. Each model
is fitted separately for individual genes, based on the
assumption that uniform scaling factors across all
genes result in inefficient normalization, particularly
for high and medium-high abundance transcripts. To
prevent overfitting, the model parameters are
regularized by pooling information across genes with
similar average expression levels. To learn robust and
smoothed parameter estimates, the Kernel regression
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
506

is applied. Finally, the true gene counts are calculated
as Pearson residuals. An improved version of the
method was chosen (Choudhary and Satija, 2022)
which excludes low-expressed genes from
regularization.
Table 2: Summary of applied scRNA-Seq data
normalization along with R package used.
Method Source Ver.
log2 - -
Freeman-
Tuke
y
- -
SCtransform
https://cran.r-
project.org/web/packages/Se
urat/index.html
5.1.0
scran
https://bioconductor.org/pac
kages/devel/bioc/vignettes/s
cran/inst/doc/scran.html
1.32.0
Dino
https://www.bioconductor.or
g/packages/release/bioc/html
/Dino.html
1.10.0
SCnorm
https://github.com/rhondaba
cher/SCnor
m
1.26.0
2.2.6 Normalized Transcript Post-
Processing
After applying all normalization methods, only the
top 20% of genes with the highest between-cells
normalized transcript variance was left to potentially
filter out non-differentially expressed ones. Next,
principal component analysis was performed and the
first 50 principal components were taken for further
analysis to reduce background noise of data.
2.3 Unsupervised Learning
For normalized and filtered data the following
unsupervised learning techniques were applied: (i)
two dimensionality reduction methods, and (ii) three
clustering methods.
2.3.1 Dimensionality Reduction
The first method was t-distributed stochastic neighbor
embedding (tSNE) (Van der Maaten and Hinton,
2008). At first, similarities between data points are
estimated (Euclidean distance here) and then
transformed into probabilities using Gaussian kernel
(high-dimensional space). Next, the low-dimensional
space is randomly generated for which each data
point has an assigned position. Similarly, the pairwise
similarities between data points are computed but
with the usage of t-distribution. The goal of tSNE is
to minimize the divergence between the pairwise
similarities in the high-dimensional space and
corresponding similarities in the low-dimensional
space. This procedure allows to preserve local
relationships and clusters within the data.
The second applied procedure was Uniform
Manifold Approximation and Projection (UMAP)
(McInnes, 2018). At first, pairwise similarities
between data points are calculated using specified
metrics (Euclidean metric here). Next, local
neighborhood structure is created based on pairwise
similarities. The optimization process between, a
random low-dimensional embedding and high-
dimensional structure is done by stochastic gradient
descent which minimizes the discrepancy between
the pairwise similarities of spaces. Moreover, the
UMAP procedure allows to preserve not only the
local relationships like tSNE but also the global ones
by constructing graph representation based on the
low-dimensional embedding (updated in iterations)
2.3.2 Clustering Algorithms
To evaluate the influence of normalization on
clustering outcomes, several common methods were
chosen. For each method, the Euclidean distance
metric was used. The optimal number of clusters was
determined by maximizing the Silhouette Index (SI)
value (Rousseeuw, 1987), calculated as the mean of
the Silhouette values computed for the entire dataset.
The first method used in this paper was k-means
(MacQueen, 1967). The main idea behind k-means is
to group observations into k pre-defined clusters,
minimizing the overall distance of each point to the
centroid of its respective cluster. When the
observations are assigned to each cluster, the
centroids are recalculated iteratively, until the loss
function reaches a plateau. Since the algorithm begins
with random initial conditions (where the preliminary
centroids are chosen from the data points), it may
produce non-deterministic outcomes. Therefore, to
find the optimal solution, it is recommended to run
the algorithm multiple times for the same pre-defined
value of k.
The second approach was hierarchical clustering
(h-clust). Specifically, agglomerative, complete-
linkage h-clust was employed, where clusters with the
smallest between-cluster distance are iteratively
combined into larger groups. This process continues
until all objects are grouped into a single cluster. In
the complete-linkage form, the between-cluster
distance is measured between the two furthest points
of each cluster (Hubert, 1974).
As the third method, the Louvain community
detection approach was used (Blondel et al., 2008).
Assessing the Influence of scRNA-Seq Data Normalization on Dimensionality Reduction Outcomes
507

Here, each cell is considered as a node and initially
assigned to its cluster. The algorithm iterates through
each node in the network, calculating the change in
modularity that would result from moving the node to
each of its neighboring clusters. If the modularity
increases, the node is merged with the cluster. This
step is repeated as long, as the increase in modularity
is no further observed. Then, the clusters are
aggregated, creating a set of new meta-communities,
and forming the nodes of a new network. The weights
of links between these meta-communities are
calculated as the sum of the weights of links between
the nodes in the corresponding original clusters. The
process is sequentially repeated, until the modularity
reaches its maximum and no further changes in
community structure occur. Before clustering, a
graph structure using the k-nearest neighbors
algorithm was constructed. To find the optimal
number of clusters, k was changed within the range 5-
100 with a step equal to 5, and the resolution
parameter from 0.4 to 2, with a step equal to 0.1.
2.4 Performance Metrics Used in
Evaluation
The silhouette index was used to estimate the effect
of normalization on dispersion after dimensionality
reduction. The index was also calculated for original
labels for comparison. The second evaluation relied
on clustering performance itself. For that, the
Adjusted Rand Index (ARI, Rand, 1971), Dice-
Sørensen coefficient (Dice, 1945; Sørensen, 1948),
and Mutual Information (Shannon, 1948) measures
were calculated.
Kruskal-Wallis test (Kruskal and Wallis, 1952)
was applied with Conover post-hoc (Conover and
Iman, 1979) to assess the difference in clustering
performance between normalization techniques. The
significance level was set to α=0.05. Additionally, the
effect size was measured using Cohen’s d modified
Conover’s d coefficient to support our inference.
During the final analysis, clustering and
dimensionality reduction methods were compared for
the same measures and statistical tests as in the
clustering performance evaluation. However, to test
differences between tSNE and UMAP the Wilcoxon
test was used (Wilcoxon, 1945).
All testing was conducted on the same PC with the
following parameters: Intel Core i5-10500 CPU @
3.10 GHz, and 64 GB of RAM. For all calculations,
computational time was collected and evaluated
alongside other performance metrics to ensure
comprehensive analysis. Furthermore, if parallel
computation was enabled within the implemented
functions, the number of cores to utilize was set to the
maximum available.
3 RESULTS
3.1 Effect of Normalization on Data
Dispersion in Reduced Space
For the BC_dis dataset reduced using tSNE, the
Kruskal-Wallis test indicated a significant difference
between methods (Figure 1A). Post-hoc Conover
tests revealed statistically significant differences
compared to non-normalized data for log2
normalization (p-value < 5.5e-6) and scran
normalization (p-value < 1.8e-9). Interestingly, FT
transformation achieved significance only before the
Bonferroni correction (uncorrected p-value = 0.03;
corrected p-value = 0.67). Conover's d effect sizes
suggest moderate effects for log2 (d = 0.47) and scran
(d = 0.59), while FT normalization exhibited a small
effect (d = 0.21) (Figure 2). In contrast, when using
UMAP for dimensionality reduction, the Kruskal-
Wallis test yielded insignificant results (Figure 1A)
marking a drastic change in findings between
reduction methods.
For the BC_sub dataset reduced using tSNE, the
Kruskal-Wallis test revealed highly significant
differences (p-value < 2.2e-16, Figure 1B). Pairwise
Conover tests confirmed significant differences for
FT (p-value < 1.3e-29), log2 (p-value < 2.8e-47), and
scran (p-value < 1.2e-50) normalizations, with large
effect sizes (d = 1.07, d = 1.37, and d = 1.42,
respectively, Fig 2). The other normalization
techniques yielded relatively small effect sizes. When
UMAP was used for dimensionality reduction, the
Kruskal-Wallis test results remained significant, but
the Conover pairwise comparisons revealed even
greater significance for FT, log2, and scran
normalizations, with corresponding effect sizes of d =
1.25, d = 1.48, and d = 1.78, respectively (Figure 2).
For the PBMC dataset, the Kruskal-Wallis test
produced significant results regardless of the
dimensionality reduction method (Figure 1C).
Pairwise multiple comparisons revealed significant
outcomes for all normalization techniques except
SCnorm. However, large effect sizes were observed
only for FT, log2, and scran normalizations. Under
tSNE, the effect sizes were d = 0.98, d = 1.04, and d
= 1.14, respectively, while UMAP yielded slightly
different effect sizes of d = 0.84, d = 1.14, and d =
1.09, respectively (Figure 2).
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
508
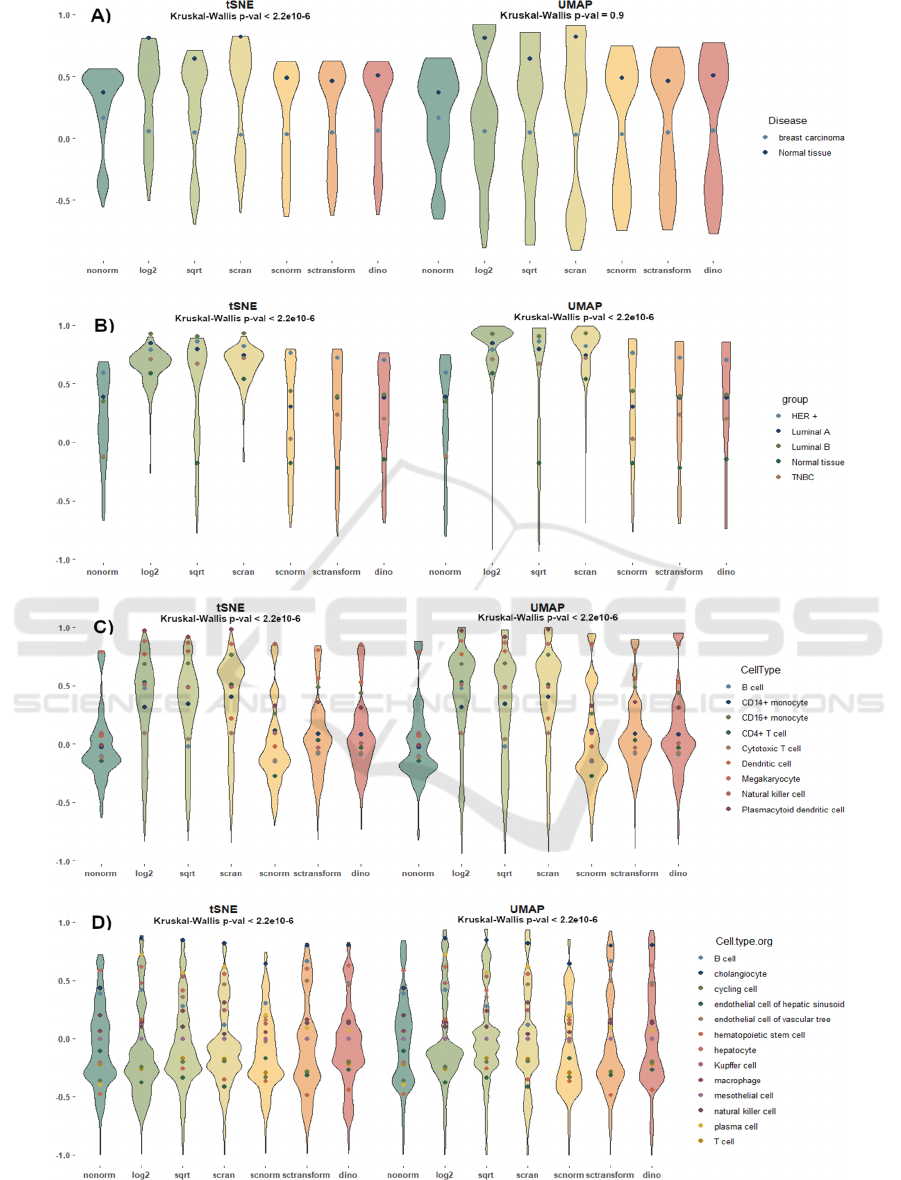
Figure 1: The violin plots illustrate the distribution of SI values after dimensionality reduction with t-SNE (left) and UMAP
(right) across all normalizations. Each point represents the mean SI values calculated across cell types. The panels show
results for A) PBMC, B) breast cancer disease, C) breast cancer subtypes, and D) liver datasets.
Assessing the Influence of scRNA-Seq Data Normalization on Dimensionality Reduction Outcomes
509

Figure 2: Results of multiple pairwise comparison. The color indicates p-value ranges after Bonferroni's correction, while the
values inside the boxes indicate the Conover’s d effect size.
In contrast to the other datasets, the Liver dataset
showed variation in outcomes depending on the
dimensionality reduction method used, despite the
Kruskal-Wallis test remaining significant overall
(Figure 1D). For tSNE, neither scran nor FT methods
reached significance, with the highest effects
observed for log2 (d = 0.25), SCnorm (d = 0.11), and
SCtransform (d = 0.09). When UMAP was applied,
log2 and scran normalizations failed to achieve
significant results. The greatest, even relatively small
effect sizes, were observed for SCnorm (d = 0.33),
SCtransform (d = 0.22), and FT (d = 0.11) (Figure 2).
These differences underscore the influence of the
dimensionality reduction method on the results.
3.2 Clustering Performance after
Normalization with Different
Methods
In BC_dis, for both ARI and MI, log2 and scran
normalization techniques outperformed all other
methods, particularly when applied in combination
with tSNE. FT transformation demonstrated better
performance than scran in scenarios where UMAP
was utilized. Overall, log2 and scran normalization
enabled the achievement of the best results for k-
means clustering, especially when paired with tSNE
(Figure 3A). For Dice and SI metrics, all techniques
produced relatively similar results, but slight
improvements were observed with Dice when
combined with tSNE, while UMAP yielded
noticeably better outcomes for SI.
For the BC_sub, when combined with tSNE, the
results across various clustering methods consistently
demonstrated the superior performance of both log2
and scran normalizations. In contrast, when paired
with UMAP, FT normalization performed slightly
better than scran. Furthermore, these normalization
techniques enabled k-means clustering to outperform
the other clustering methods (Figure 3B).
In PBMC, according to Dice values, the weakest
outcomes were observed with log2, FT, and scran
normalizations, while the best results were achieved
using tSNE combined with k-means clustering
(Figure 3C). In the case of MI values, and for Louvain
clustering, all normalization methods, except
SCnorm, demonstrated relatively better performance
compared to non-normalized data. In an overall
comparison, log2, FT, and scran normalization
methods outperformed the others. In Louvain
clustering, the choice of dimensionality reduction
method did not significantly impact the results,
except for dino, which showed a marked
improvement when combined with UMAP. It is
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
510
worth mentioning that SCransform followed by
UMAP and clustering with either h-clust or k-means
yielded results even worse than non-normalized data.
ARI results consistently highlighted the advantages
of log2, FT, and scran normalization techniques,
particularly when combined with tSNE and k-means
clustering. In contrast to non-normalized data, SI
values showed little to no improvement when reduced
with tSNE across all clustering methods. However,
the scenario drastically changed when UMAP was
used. Here, improvements were observed across all
normalizations, with notable gains seen in k-means
and h-clust, especially with FT, log2, scran, and dino
techniques.
In contrast to the previous datasets, in liver for
tSNE reduction, only SCnorm normalization led to
improved ARI values compared to non-normalized
data - and this improvement was observed exclusively
after applying k-means clustering (Figure 3D). For
UMAP, small improvements were noted with
SCtransform, while the other normalization methods
resulted in performance deterioration. Similar trends
were observed for Dice coefficient values. For MI
values, when tSNE was used, all normalization
methods led to slight improvements. However, log2,
FT, and scran showed marginally better performance
compared to the others. When UMAP was applied,
the results improved across all normalization
methods, with the best outcomes achieved using the
same methods as in tSNE. It was observed that the
performance of SCnorm varied depending on the
dimensionality reduction method and clustering
technique. SCnorm performed worse with tSNE but
showed significantly better performance with UMAP
when followed by h-clust. Similarly, dino
normalization performed substantially better with
tSNE but slightly worse with UMAP when followed
by k-means clustering. The SI values were relatively
poor, similar to those observed with the PBMC
dataset. However, overall performance improved
when the data was reduced using UMAP.
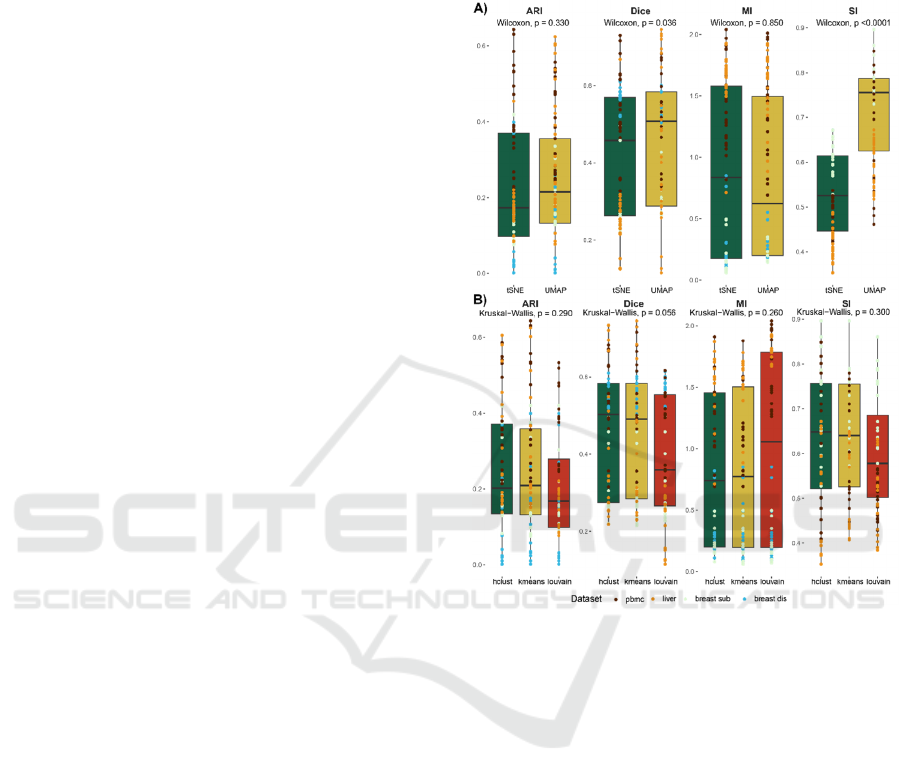
3.3 Unsupervised Method Impact
We observed differences in clustering efficiency
across the same normalization methods with varying
dimensionality reduction methods. Therefore, the
results were compared between these reduction
methods, with particular attention to the clustering
metrics utilized in this study. The results of Wilcoxon
testing (Figure 4A) showed no statistically significant
differences for both ARI and MI metrics. Although,
such differences exist for Dice and SI. Kruskal-Wallis
test did not reveal statistically significant differences
between clustering methods for any of the approaches
used (Figure 4B), though, Louvain performed slightly
worse compared to both k-means and h-clust.
Figure 3: Results evaluated on aggregated datasets. Panel
A) represents a comparison of dimensionality reduction
methods, without distinguishing between clustering
methods. Panel B) represents a comparison of clustering
method without division between dimensionality reduction.
For each comparison, the corresponding Wilcoxon’s test p-
values are indicated.
3.4 Computational Time
Finally, we investigated computational time of tested
normalizations (Table 3) as well as dimensionality
reduction methods (Table 4). As can be observed all
normalizations computational time increase with the
increasing number of samples/cells in experiment. As
expected the simplest mathematical procedures were
the fastest i.e. the log2 and FT normalization. Next
scran and SCtransform can be distinguished. The
worst time performance was observed for
SCnorm. A
similar trend was observed for dimensionality reduction
methods, however, UMAP significantly outperformed
tSNE in terms of computational time.
Assessing the Influence of scRNA-Seq Data Normalization on Dimensionality Reduction Outcomes
511
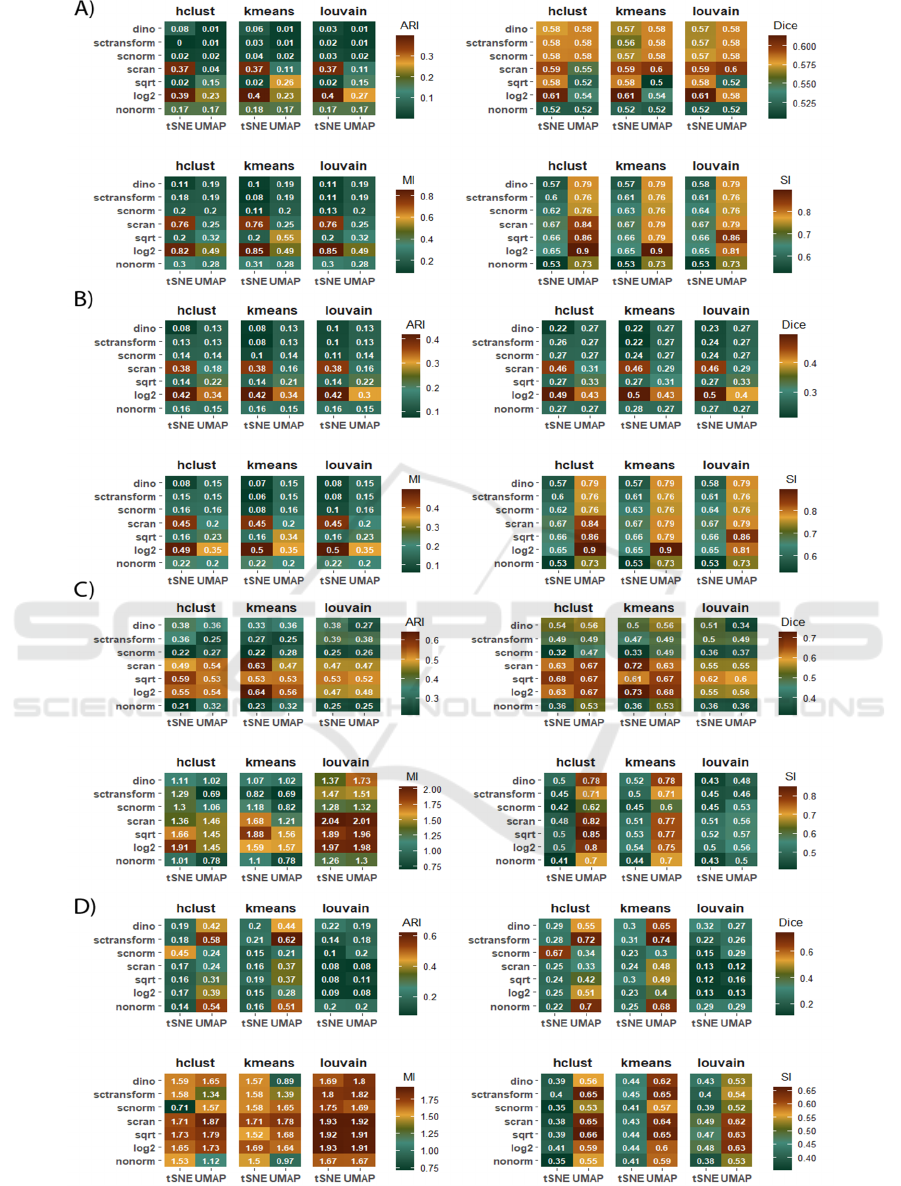
Figure 4: Comparison of clustering efficiency measures calculated with distinction to both normalization and dimensionality
reduction methods. Each subplot presents metric values for ARI, Dice, MI, and SI, arranged from top left to bottom right.
Subsequent subplots show results for A) breast cancer disease, B) breast cancer subtypes, C) PBMC, and D) liver datasets.
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
512

Table 3: Computational time of normalization methods (in
seconds).
Dataset Liver PBMC
Breast
Cancer
log2 12.31 2.02 0.25
Freeman-Tukey 6.46 0.58 0.18
SCtransform 117.33 25.55 7.00
scran 93.72 17.55 2.70
Dino 2431.92 459.36 295.98
SCnorm 15850.23 3682.53 7640.03
Table 4: Computational time of dimensionality reduction
methods by average across normalizations (in seconds).
Dataset Liver PBMC Breast Cancer
UMAP 25.06 10.71 1.78
t-SNE 474.84 105.82 0.89
4 DISCUSSION AND
CONCLUSIONS
This study thoroughly examined the impact of
specific data normalization methods on the efficiency
of scRNA-seq data downstream analysis. Our results
indicate that simple normalization methods, such as
log2 and scran consistently enabled obtaining
superior outcomes compared to scRNA-seq domain-
specific techniques, especially for the small and
medium-sized datasets. Moreover, depending on the
dimensionality reduction method leveraged in the
processing, FT normalization sometimes obtains
superior performance compared to scran. However,
as dataset size increases, the performance gap
between simple normalizations and scRNA-seq-
specific techniques diminishes. This observation
suggests that for large and extra-large datasets,
specialized normalization techniques may become
essential to achieve optimal results. On the other
hand, the SCnorm, SCtransform, and dino techniques
appear to be sensitive to specific steps within the
overall pipeline procedure, a trend particularly
noticeable with the larger, Liver dataset. Therefore,
we strongly recommend to be aware when deciding
whether to use simple or domain-specific techniques.
It is worth noting that, in addition to overall
outcomes and performance measures, domain-
specific techniques demand significantly more
computing time. SCnorm normalization, in particular,
is better suited for smaller datasets, as its
computational requirements increase drastically with
larger datasets containing thousands of cells. Similar
conclusions also occur in the literature (Zhang et al.,
2023). Consequently, its inferior performance
compared to other methods, especially for smaller
datasets, is particularly surprising. The observed
dependence was made on few datasets and large scale
research is still needed.
In the overall comparison, it was noticed, that for
a smaller breast cancer dataset, tSNE enabled to
achieve slightly better clustering outcomes than
UMAP, regardless of the level of cell-type
differentiation. On the other hand, UMAP achieved
even statistically significant better results within SI
metric. However, these differences likely arise from
the specific manner in which UMAP performs
dimensionality reduction. Finally, the type of
normalization technique used before reduction may
affect the final level of data dispersion. Yet, presented
research does not include all dimensionality reduction
techniques like variational autoencoder (VAE) or
SIMLR (Wang et al., 2017) which were teste in
(Xiang et al., 2021) but not in terms of normalization
impact. Next, for both tested dimensionality
reduction techniques the impact of distance metric
might be as well observed. In presented research only
Euclidian distance was considered in UMAP and
tSNE.
Furthermore, compared to Louvain clustering, the
superior performance of k-means and h-clust was
consistently observed. However, statistical inference
did not reveal statistically significant differences for
any clustering metric across the clustering methods
evaluated.
Summarizing the results, it is evident that the
choice of normalization technique depends on the size
and diversity of the dataset, as different methods can
produce varying outcomes. Simple normalization
techniques, like log2 and FT, despite not accounting
for the complexity of the scRNA-seq data
characteristics, still yielded relatively good results.
Therefore, careful planning of the scRNA-seq data
processing pipeline is crucial, as each particular step
can strongly affect the final analysis outcomes.
ACKNOWLEDGMENTS
This study was supported by the Silesian University
of Technology grant for maintaining and developing
research potential [MO, MM] and the Excellence
Initiative - Research University program
implemented at the Silesian University of
Technology no. 02/070/SDU/10-21-01 [JZ].
Assessing the Influence of scRNA-Seq Data Normalization on Dimensionality Reduction Outcomes
513

REFERENCES
Ahlmann-Eltze, C., & Huber, W. (2023). Comparison of
transformations for single-cell RNA-seq data. Nature
Methods, 20(5), 665-672.
Bacher, R., Chu, L. F., Leng, N., Gasch, A. P., Thomson, J.
A., Stewart, R. M., Newton, M., & Kendziorski, C.
(2017). SCnorm: Robust normalization of single-cell
RNA-seq data. Nature Methods, 14(6), 584–586.
Blondel, V. D., Guillaume, J.-L., Lambiotte, R., &
Lefebvre, E. (2008). Fast unfolding of communities in
large networks. Journal of Statistical Mechanics:
Theory and Experiment, 2008(10), P10008.
Booeshaghi, A. S., Hallgrímsdóttir, I. B., Gálvez-Merchán,
Á., & Pachter, L. (2022). Depth normalization for
single-cell genomics count data. bioRxiv.
Brown, J., Ni, Z., Mohanty, C., Bacher, R., & Kendziorski,
C. (2021). Normalization by distributional resampling
of high throughput single-cell RNA-sequencing data.
Bioinformatics, 37(22), 4123-4128.
Cakir, B., Prete, M., Huang, N., Van Dongen, S., Pir, P., &
Kiselev, V. Y. (2020). Comparison of visualization
tools for single-cell RNAseq data. NAR Genomics and
Bioinformatics, 2(3), lqaa052.
Choudhary, S., & Satija, R. (2022). Comparison and
evaluation of statistical error models for scRNA-seq.
Genome Biology, 23(1), 27.
Chung, W., Eum, H. H., Lee, H. O., Lee, K. M., Lee, H. B.,
Kim, K. T., ... & Park, W. Y. (2017). Single-cell RNA-
seq enables comprehensive tumour and immune cell
profiling in primary breast cancer. Nature
communications, 8(1), 15081.
Cole, M. B., Risso, D., Wagner, A., DeTomaso, D., Ngai,
J., Purdom, E., ... & Yosef, N. (2019). Performance
assessment and selection of normalization procedures
for single-cell RNA-seq. Cell systems, 8(4), 315-328.
Conover, W. J., & Iman, R. L. (1979). On multiple-
comparisons procedures (Tech. Rep. LA-7677-MS).
Los Alamos Scientific Laboratory.
Cuevas-Diaz Duran, R., Wei, H., & Wu, J. (2024). Data
normalization for addressing the challenges in the
analysis of single-cell transcriptomic datasets. BMC
Genomics, 25, 444.
Dice, L. R. (1945). Measures of the amount of ecologic
association between species. Ecology, 26(3), 297–302.
Ding, J., Adiconis, X., Simmons, S. K., Kowalczyk, M. S.,
Hession, C. C., Marjanovic, N. D., ... & Levin, J. Z.
(2020). Systematic comparison of single-cell and
single-nucleus RNA-sequencing methods. Nature
biotechnology, 38(6), 737-746.
Do, V. H., & Canzar, S. (2021). A generalization of t-SNE
and UMAP to single-cell multimodal omics. Genome
Biology, 22(1), 130.
Hafemeister, C., & Satija, R. (2019). Normalization and
variance stabilization of single-cell RNA-seq data using
regularized negative binomial regression. Genome
Biology, 20(1), 296.
Hubert, L. (1974). Approximate evaluation techniques for
the single-link and complete-link hierarchical
clustering procedures. Journal of the American
Statistical Association, 69(347), 698–704.
Hwang, B., Lee, J. H., & Bang, D. (2018). Single-cell RNA
sequencing technologies and bioinformatics pipelines.
Experimental & molecular medicine, 50(8), 1-14.
Kruskal, W. H., & Wallis, W. A. (1952). Use of ranks in
one-criterion variance analysis. Journal of the
American Statistical Association, 47(260), 583–621.
Lause, J., Berens, P., & Kobak, D. (2021). Analytic Pearson
residuals for normalization of single-cell RNA-seq
UMI data. Genome Biology, 22(1), 258.
Luecken, M. D., & Theis, F. J. (2019). Current best
practices in single ‐ cell RNA
‐ seq analysis: A
tutorial. Molecular Systems Biology, 15(6).
Lun, A. T. L., Bach, K., & Marioni, J. C. (2016). Pooling
across cells to normalize single-cell RNA sequencing
data with many zero counts. Genome Biology, 17(1).
Lytal, N., Ran, D., & An, L. (2020). Normalization methods
on single-cell RNA-seq data: an empirical survey.
Frontiers in genetics, 11, 41.
MacQueen, J. B. (1967). Some methods for classification
and analysis of multivariate observations. In L. M. Le
Cam & J. Neyman (Eds.), Proceedings of the fifth
Berkeley symposium on mathematical statistics and
probability (Vol. 1, pp. 281–297). University of
California Press.
Marczyk, M., Jaksik, R., Polanski, A., & Polanska, J.
(2018). Gamred—Adaptive filtering of high-
throughput biological data. IEEE/ACM Transactions
on Computational Biology and Bioinformatics, 17(1),
149-157.
McInnes, L., Healy, J., Saul, N., & Großberger, L. (2018).
UMAP: Uniform Manifold Approximation and
Projection. Journal of Open Source Software, 3(29).
Rand, W. M. (1971). Objective criteria for the evaluation of
clustering methods. Journal of the American Statistical
Association, 66(336), 846-850.
Rousseeuw, P. J. (1987). Silhouettes: a graphical aid to the
interpretation and validation of cluster analysis. Journal
of computational and applied mathematics, 20, 53-65.
Shannon, C. E. (1948). A mathematical theory of
communication. Bell System Technical Journal, 27(3),
379–423.
Sørensen, T. (1948). A method of establishing groups of
equal amplitude in plant sociology based on similarity
of species and its application to analyses of the
vegetation on Danish commons. Kongelige Danske
Videnskabernes Selskab, 5(4), 1–34.
Tukey, J. W. (1949). Comparing individual means in the
analysis of variance. Biometrics, 99-114.
Vallejos, C. A., Risso, D., Scialdone, A., Dudoit, S., &
Marioni, J. C. (2017). Normalizing single-cell RNA
sequencing data: challenges and opportunities. Nature
methods, 14(6), 565-571.
Van der Maaten, L., & Hinton, G. (2008). Visualizing data
using t-SNE. Journal of machine learning research,
9(11).
Vieth, B., Parekh, S., Ziegenhain, C., Enard, W., &
Hellmann, I. (2019). A systematic evaluation of single
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
514

cell RNA-seq analysis pipelines. Nature
communications, 10(1), 4667.
Wang, Z. Y., Keogh, A., Waldt, A., Cuttat, R., Neri, M.,
Zhu, S., ... & Nigsch, F. (2021). Single-cell and bulk
transcriptomics of the liver reveals potential targets of
NASH with fibrosis. Scientific reports, 11(1), 19396.
Wang, B., Zhu, J., Pierson, E., Ramazzotti, D., and
Batzoglou, S. (2017). Visualization and analysis of
single-cell RNA-seq data by kernel-based similarity
learning. Nat. Methods 14, 414–416.
Wilcoxon, F. (1945). Individual comparisons by ranking
methods. Biometrics Bulletin, 1(6), 80–83.
Xiang, R., Wang, W., Yang, L., Wang, S., Xu, C., & Chen,
X. (2021). A comparison for dimensionality reduction
methods of single-cell RNA-seq data. Front. Genet. 12,
646936.
Zhang, S., Li, X., Lin, J., Lin, Q., & Wong, K. C. (2023).
Review of single-cell RNA-seq data clustering for cell-
type identification and characterization. RNA, 29(5),
517–530.
Assessing the Influence of scRNA-Seq Data Normalization on Dimensionality Reduction Outcomes
515
