
PathDisGene: Discovering Informative Gene Groups for Disease
Diagnosis Using Pathway-Disease Associations and a Grouping,
Scoring, Modeling-Based Machine Learning Approach
Emma Qumsiyeh
1a
, Burcu Bakir-Gungo
2b
and Malik Yousef
2c
1
Faculty of Engineering and Information Technology, Palestine Ahliya University, Bethlehem, Palestine
2
Department of Computer Engineering, Faculty of Engineering, Abdullah Gul University, Kayseri, Turkey
Keywords: Grouping-Scoring-Modeling (G-S-M) Approach, Machine Learning, Biological Integrative Approach,
Feature Selection, Pathway-Disease Associations, Comparative Toxicogenomics Database (CTD),
Biomarkers.
Abstract: Recently, machine learning and various feature selection techniques have become popular for understanding
the relationship between genes, molecular pathways, and diseases. Integrating existing domain knowledge
into biological data analysis has demonstrated considerable potential for finding new biomarkers with
translational uses. This paper presents PathDisGene, an innovative machine-learning tool that integrates
existing domain knowledge by utilizing a Grouping-Scoring-Modeling (G-S-M) approach to discover
associations among gene-pathway-disease. The first step in PathDisGene is the grouping component that
associates genes according to their biological associations with diseases and pathways. This component uses
the Comparative Toxicogenomics Database (CTD). Subsequently, the scoring component is applied to score
each group and the highest-ranked groupings are then used to train the classifier. We test PathDisGene on ten
GEO datasets and demonstrate its performance, where most of them are with high accuracy, sensitivity,
specificity, and AUC values across various diseases. The tool's capacity to recognize new pathway-disease
associations and uncover connections between pathways and diseases along their associated genes
underscores its potential as a significant asset in promoting precision medicine and systems biology.
1 INTRODUCTION
Complex diseases are caused by a combination of
genetic factors and environmental effects. Since they
do not follow any patterns of inheritance, research
efforts are conducted to discover various disease
biomarkers (MacEachern & Forkert, 2021). Most of
the research in this field focused on gene expression
patterns. They seek to identify disease-associated
genes that may function as biomarkers for early
diagnosis, prognosis, and the formulation of targeted
therapy approaches. Identifying biomarkers and
classifying samples have become essential domains
in bioinformatics research (MacEachern & Forkert,
2021).
Treating complex human diseases increasingly
relies on accurate patient stratification facilitated by
a
https://orcid.org/0000-0002-3797-5851
b
https://orcid.org/0000-0002-2272-6270
c
https://orcid.org/0000-0001-8780-6303
bio-indicators obtained from genomics,
transcriptomics, and proteomics. Traditional feature
selection methods frequently neglect the relationships
among features, concentrating solely on the
significance of individual genes. However, one
should consider that genes act together as part of a
group at genomic levels. Enhanced insights can be
achieved when tools leverage biological information
for comprehensive analysis rather than relying solely
on traditional clustering and machine-learning
techniques (Holzinger et al., 2017).
Gene-pathway-disease associations are complex
relations. Genes, the fundamental units of genetics,
encode proteins that sustain cellular homeostasis and
enable intercellular communication. Disease states
frequently arise from genetic abnormalities or
dysregulations that limit these mechanisms. Cancers
676
Qumsiyeh, E., Bakir-Gungo, B. and Yousef, M.
PathDisGene: Discovering Informative Gene Groups for Disease Diagnosis Using Pathway-Disease Associations and a Grouping, Scoring, Modeling-Based Machine Learning Approach.
DOI: 10.5220/0013378200003911
Paper published under CC license (CC BY-NC-ND 4.0)
In Proceedings of the 18th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2025) - Volume 1, pages 676-683
ISBN: 978-989-758-731-3; ISSN: 2184-4305
Proceedings Copyright © 2025 by SCITEPRESS – Science and Technology Publications, Lda.
often arise from genetic anomalies that lead to
unregulated cell proliferation resulting from mistakes
in cell division mechanisms (Łukasiewicz et al.,
2021). Biological pathways are sequential molecular
processes within cells that induce specific cellular
alterations. They affect various biological functions,
including metabolism, gene expression, and cellular
signaling. Dysregulation of pathways, such as the
MAPK signaling system, regulates cell proliferation
and differentiation, and it can result in severe health
conditions, including cancer (Jin et al., 2014).
Recent advancements in the field of bioinformatics
have been accelerated by easy access to extensive
datasets and comprehensive repositories such as Gene
Expression Omnibus (GEO) (Barrett et al., 2013),
miRTarBase (Hsu et al., 2011), the Cancer Genome
Atlas (TCGA) (Tomczak et al., 2015), and the
Comparative Toxicogenomics Database CTD (Davis
et al., 2021). These databases facilitate researchers in
validating ideas in silico and employing machine
learning to uncover biomarkers to classify diseases.
Integrating this knowledge while building machine
learning can enhance the prediction task.
Yousef et al. developed the Grouping-Scoring-
Modeling (G-S-M) methodology for the integration of
biological knowledge utilizing numerous
computational tools, including maTE (Yousef et al.,
2019), CogNet (Yousef, Ülgen, et al., 2021),
mirCorrnet (Yousef, Goy, et al., 2021) and PriPath
(Yousef, Ozdemir, et al., 2022). The integration of
biological knowledge with gene expression selection
was examined in SVM-RCE-R; the initial report
focused on groups of genes rather than individual
genes (Yousef, Bakir-Gungor et al., 2021). SVM-RCE
(Support Vector Machines - Recursive Cluster
Elimination) categorizes genes based on their
expression values and evaluates each gene cluster
using a machine-learning algorithm (Yousef, Jabeer, et
al., 2021). In a recent work, Yousef et al. utilized Gene
Ontology terms and the G-S-M model for gene
expression data analysis (Ersoz et al., 2023). Besides,
it has been used to detect molecular subtypes in BRCA
(Qumsiyeh, Bakir-Gungor, et al., 2024) and to rescore
multiple groups using different machine learning
algorithms (Qumsiyeh, Yousef, et al., 2024). This
study primarily utilizes the G-S-M methodology to
categorize genes and identify the most relevant groups
associated with a pathway-disease association.
PathDisGene, our innovative machine learning
framework, employs a Grouping-Scoring-Modeling
(G-S-M) approach that groups genes by integrating
biological knowledge about pathway-disease
associations from the Comparative Toxicogenomics
Database (CTD) database. In Monte Carlo cross-
validation (MCCV), random sample subsets are
considered as the training dataset, while the
remaining samples are allocated to the testing dataset.
In each training iteration, the most informative
pathway-disease-gene groups are determined, and
subsequently, the cumulatively top-ranked groups are
used to train the model.
PathDisGene aims not to compete with previously
published tools targeting single-disease markers but
rather to identify new gene clusters associated with
several pathways and diseases. Utilizing a G-S-M
strategy, PathDisGene improves comprehension of
pathway-disease associations, facilitating novel
diagnostic and therapeutic advancements.
2 DATASETS
2.1 GEO Dataset
We downloaded 10 human gene expression datasets
for different complex diseases from the GEO
database (Barrett et al., 2013). For each dataset, we
specified the GEO accession, the name of the disease,
and the number of positive and negative samples. The
characteristics of the 10 datasets are presented in
detail in Table 1.
Table 1: Description of the 10 GEO datasets used in
PathDisGene.
GEO
Accession
Title
#Sample
s
Classes
GDS1962
Glioma-derived stem cell
factor effect on
angiogenesis in the brain
180
Negative = 23,
Positive = 157
GDS2545
Metastatic prostate
cancer (HG-U95A)
171
Negative = 81,
Positive = 90
GDS2771
Large airway epithelial
cells from cigarette
smokers with suspected
lung cance
r
192
Negative = 90,
Positive = 102
GDS3257
Cigarette smoking effect
on lung adenocarcinoma
107
Negative = 49,
Positive = 58
GDS4206
Pediatric acute leukemia
patients with early
relapse: white blood cells
197
Negative =
157, Positive =
40
GDS5499
Pulmonary hypertension:
PBMCs
140
Negative = 41,
Positive = 99
GDS3837
Non-small cell lung
carcinoma in female
nonsmokers
120
Negative = 60,
Positive = 60
GDS4516
_4718
Colorectal cancer: laser
microdissected tumor
tissues
148
Negative = 44,
Positive = 104
GDS2547
Metastatic prostate
cancer (HG-U95C)
164
Negative = 75,
Positive = 89
GDS3268
Colon epithelial biopsies
of ulcerative colitis
patients
202
Negative = 73,
Positive = 129
PathDisGene: Discovering Informative Gene Groups for Disease Diagnosis Using Pathway-Disease Associations and a Grouping, Scoring,
Modeling-Based Machine Learning Approach
677

2.2 Pathway-Disease Associations
We have downloaded the disease-pathway
associations dataset from the Comparative
Toxicogenomics Database (CTD). CTD is a
comprehensive, publicly accessible resource
designed to enhance understanding of how
environmental exposures impact human health. By
providing curated information on chemical–
gene/protein interactions, chemical–disease, and
gene-disease relationships, CTD integrates these data
with functional and pathway insights, supporting
hypothesis generation about the mechanisms driving
environmentally influenced diseases.
The dataset includes key fields such as
DiseaseName, DiseaseID, PathwayName,
PathwayID (linked to KEGG or REACTOME
identifiers), and InferenceGeneSymbol, which
denotes the gene through which the association is
inferred. We adopted a novel approach by integrating
disease and pathway information into a single group
column. This structure differs from the traditional
format used in the CTD Database. By combining the
disease and pathway columns into a single group
column, we streamlined the representation of disease-
pathway associations. This unified format facilitates
the direct mapping of diseases to their respective
pathways and genes. After processing the dataset,
76,966 unique disease_pathway associations were
found. Besides, there are 4,388 unique genes, 317
unique pathways, and 3,176 unique diseases.
3 METHODOLOGIES
PathDisGene is a novel approach built on the basic
concepts of the Grouping-Scoring-Modeling (G-S-
M) approach (Yousef et al., 2024). This framework
combines machine learning capabilities with
comprehensive biological knowledge to identify
groups of genes or features. PathDisGene groups
these genes or features into biological groups and
ranks those groups based on their contribution to the
target class in a two-class dataset, such as a diseased
condition versus a normal condition.
Embedded feature selection is a key component of
the G-S-M approach. This procedure systematically
employs machine learning algorithms to identify the
most informative groups of features, hence increasing
the ability to distinguish between different classes. By
integrating essential biological insights, the G-S-M
framework seeks to unravel complex biological
phenomena, thereby fostering novel discoveries.
The primary goal of the G-S-M approach is to
provide a flexible framework that can be applied to
any dataset where existing biological knowledge
allows for the categorization of observable features.
This method initially requires two-class datasets and
utilizes existing biological knowledge (such as genes
related to a biological pathway) to group the data.
Each group uses a scoring process that includes
internal cross-validation and statistical approaches to
determine their importance.
PathDisGene, based on the G-S-M approach,
seeks to enhance the investigation of gene groupings
by incorporating multiple sources of biological
knowledge, such as disease-target genes, disease-
pathway associations, and pathway data.
PathDisGene is inspired by previous tools like
miRGediNET (Qumsiyeh, Salah, et al., 2023),
GediNET (Qumsiyeh et al., 2022), GediNETPro
(Qumsiyeh, Yazıcı, et al., 2023), CogNet (Yousef,
Ülgen, et al., 2021), maTE (Yousef et al., 2019),
mirCorrnet (Yousef, Goy, et al., 2021),
miRModuleNet (Yousef, Goy, et al., 2022), SVM-
RCE-R (Yousef, Bakir-Gungor, et al., 2021), PriPath
(Yousef, Ozdemir, et al., 2022), miRdisNET (Jabeer
et al., 2023), GeNetOntology (Ersoz et al., 2023), and
detecting semantic similarity (Qumsiyeh, Yousef, et
al., 2023). PathDisGene's extensive capabilities are
made possible by the foundation of the earlier tools
created to use particular biological information in
gene grouping.
3.1 PathDisGene Tool
In this study, we introduce a novel machine-learning-
based tool named PathDisGene, designed to utilize
prior biological knowledge from pre-existing
biological knowledge. The tool presents an
integrative machine learning-based approach based
on the G-S-M methodology. This approach includes
segregating data, grouping genes based on the pre-
existing biological knowledge obtained from the
CTD database, applying scoring metrics, and utilizing
machine learning techniques. The Random Forest
was considered in the Scoring and in the Modeling,
but one was also able to use other algorithms.
Random Forest classifier was used with defaults
parameters where the number of estimators is 100.
The overview of the methodological process involved
in the PathDisGene tool is presented below:
3.1.1 Initial Data Segmentation
The process starts by partitioning the dataset into two
parts: 90% for training and the remaining 10% for
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
678

testing. This partitioning is critical to ensure the tool
is trained and evaluated on distinct data sets, allowing
for an accurate assessment of its predictive
capabilities.
3.1.2 The G Grouping Component
The first step involves creating groups of genes by
integrating prior knowledge about pathway_disease
associations. The output of this process is a list of
groups, where each group consists of a set of genes.
This grouping leverages previously acquired
biological knowledge to ensure the genes are
categorized based on relevant biological
characteristics. The next step involves extracting a
sub-dataset for each group from the training part of
the dataset. In this step, the input consists of the list
of gene groups and the training data. Each sub-dataset
represents the genes in a particular group and
maintains its original class label, such as positive or
negative.
3.1.3 The S Scoring Component
The scoring component aims to assign scores to each
group, assessing the significance of the group for
classifying the data based on the genes that are
members of the group. The input to the S component
is all the two-class sub-datasets created in the G
component. We have used the Random Forest
algorithm with five randomized subsampling cross-
validation techniques to compute the score. The score
was the mean of the accuracy. Groups are ranked and
then prioritized based on their scoring outcomes. The
highest-scoring groups are chosen and moved
forward to the next step, the machine learning
modeling phase.
3.1.4 The M Model Construction
Component
This phase focuses on constructing a machine-
learning model using the gene groups that received
the highest scores in the previous step (S component).
The Random Forest classifier is utilized in this
context, and the model's performance is evaluated
using the validation dataset.
3.1.5 Iterative Assessment with Randomized
Subsampling Cross-Validation
Technique
An iterative loop of randomized subsampling cross-
validation technique, repeated 100 times, underpins
the entire PathDisGene process from data
segmentation to final model evaluation. This
repetitive approach guarantees a comprehensive and
reliable assessment, showcasing the tool's accuracy
and effectiveness.
4 EVALUATION
We employed the PathDisGene, partitioning the
data into 90% for training and 10% for testing. Due
to the imbalanced nature of the datasets, characterized
by an unequal distribution of class labels, we utilized
the under-sampling strategy. This method addresses
imbalanced datasets by preserving all samples in the
minority class while reducing the size of the majority
class. We utilized tenfold Monte Carlo cross-
validation (MCCV) (Randomized subsampling cross-
validation)(Xu & Liang, 2001) for model training. In
MCCV, parts of the samples are randomly designated
as training data, while the remainder is allocated for
testing data. The performance metrics are calculated
as the mean of 100-fold MCCV. Various quantitative
metrics are computed, including accuracy,
specificity, sensitivity, Precision, F1-measure and the
area under the receiver operating characteristic
(ROC) curve (Dalianis, 2018).
5 RESULTS & DISCUSSION
Table 2 comprehensively analyses PathDisGene's
efficacy among the top 10 gene groups in the
GDS3257 (Lung adenocarcinoma) dataset. The data
represent average values from 100 MCCV iterations,
illustrating the performance metrics for cumulative
groupings of top-ranked genes. This analysis displays
the overall performance of the highest-ranked groups
corresponding to each row in Table 2.
The initial row (# of Groups = 1) demonstrates the
performance metrics utilizing only the highest-ranked
group of genes, which has 2.71 features on average.
This initial group attained an AUC of 97%, which
signifies its exceptional discriminatory capability.
Moreover, additional performance metrics, including
sensitivity (94.8%), specificity (93.8%), and accuracy
(94.3%), further emphasize the significance of this
group.
In the second row (# of Groups = 2), the
performance metrics indicate the cumulative impact
of genes from the first and second-highest-ranked
groups, with 4.13 features on average. Compared to
including only one group, performance metrics are
significantly enhanced, with an AUC of 97.9% and an
PathDisGene: Discovering Informative Gene Groups for Disease Diagnosis Using Pathway-Disease Associations and a Grouping, Scoring,
Modeling-Based Machine Learning Approach
679
accuracy rise to 95.5%, demonstrating the beneficial
effect of including more genes.
As the cumulative number of groups rises, the
performance indicators constantly increase. For
instance, by the sixth group (# of Groups = 6), the
model attains an AUC of 99.1% and an accuracy of
96.8%, highlighting improved prediction.
Correspondingly, the sensitivity, specificity, and F-
measure metrics demonstrate consistent
improvements, reflecting balanced performance
across all principal measures.
Upon including all 10 groups (# of Groups = 10),
the model attains optimal performance, reaching an
AUC of 99.6% with an average of 12.24 features.
This underscores the model's capacity to efficiently
leverage supplementary genes to improve predictive
accuracy and overall efficacy. Metrics like sensitivity
(97.8%), specificity (96.8%), and accuracy (97.2%)
exhibit exceptional stability and repeatability, hence
reinforcing the efficacy of the cumulative approach.
The findings in Table 2 highlight that increasing
the quantity of top-ranked gene groups enhances the
performance of PathDisGene, demonstrating its
efficacy in predictive modeling for the GDS3257
dataset.
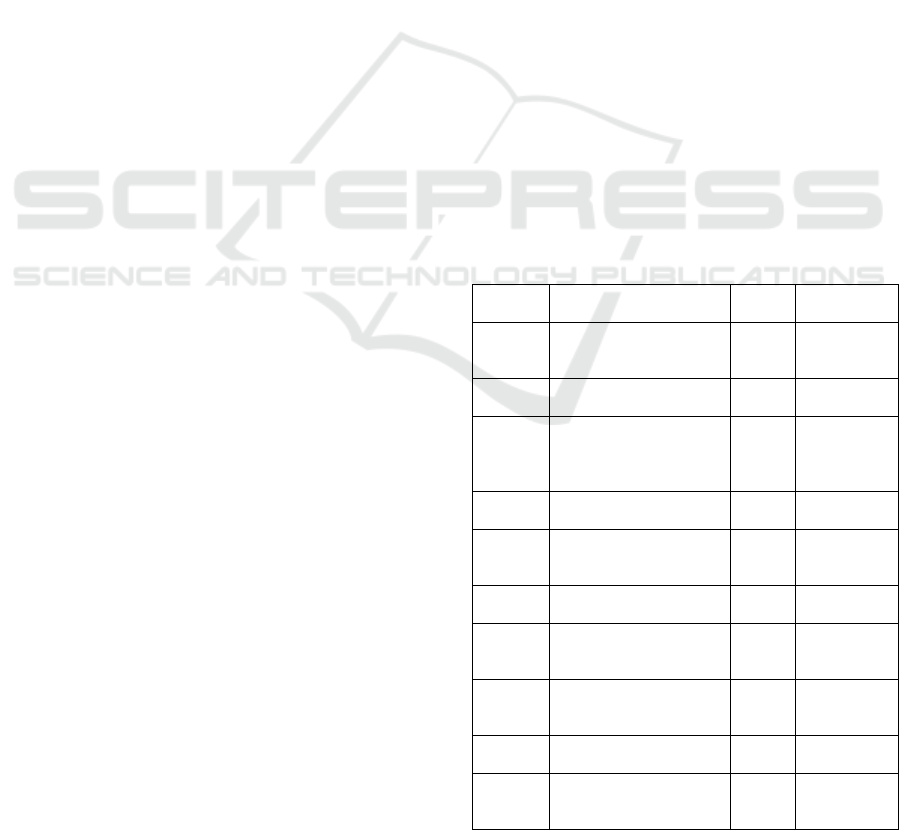
Table 2: The average Cumulative Performance of
PathDisGene across the top 10 Gene Groups in the
GDS3257 Dataset over the 100 MCCV Iterations.
# of Groups
# of Features
Sensitivity
Specificity
Precision
Accuracy
Area Under
Curve
F-measure
1 2.71 0.94 0.93 0.94 0.94 0.97 0.94
2 4.13 0.96 0.944 0.95 0.95 0.97 0.95
3 5.6 0.97 0.94 0.95 0.95 0.98 0.96
4 6.63 0.97 0.95 0.95 0.96 0.98 0.96
5 7.82 0.97 0.95 0.96 0.96 0.98 0.96
6 8.73 0.97 0.96 0.96 0.96 0.99 0.96
7 9.8 0.97 0.96 0.96 0.96 0.99 0.96
8 10.57 0.97 0.96 0.96 0.97 0.99 0.97
9 11.58 0.97 0.96 0.97 0.97 0.99 0.97
10 12.24 0.97 0.96 0.97 0.97 0.99 0.97
Table 3 presents an in-depth evaluation of
PathDisGene's efficacy across ten GEO datasets,
emphasizing the second-top-ranking groups. The
outcomes obtained from the mean of 100 MCCV
iterations include essential performance metrics such
as sensitivity, specificity, precision, accuracy, area
under the receiver operating characteristic curve, and
the F-measure. Each dataset is assessed according to
the number of features (genes) linked to the two
categories, demonstrating varying performance levels
among datasets.
The mean number of features across the datasets
is roughly 3.67, indicating diversity in genetic
representation and complexity. Among the datasets,
GDS3837 exhibits exceptional performance,
attaining a sensitivity of 87.8%, specificity of 90.5%,
accuracy of 91.2%, and an AUC of 94.2%, resulting
in a notable F-measure of 88.8%. This exceptional
performance highlights the resilience of the chosen
groupings within this dataset.
GDS1962 is notable for attaining an AUC of
93.6%, robust sensitivity (91.4%) and precision
(93.4%), and an overall accuracy of 88.4%. The
results demonstrate the dataset's capacity to facilitate
good predictive modeling with a limited number of
features (3.11 genes).
On the other hand, GDS4206 and
GDS4516_4718 present as challenging datasets,
demonstrating significantly lower performance
measures. Both datasets exhibit a sensitivity of
36.2%, accompanied by moderate specificity (77.1%)
and low precision (43.8%). The accuracy for these
datasets is 64.5%, accompanied by an AUC of 61.5%,
indicating the challenges presented by the particular
features within these datasets. However, it is worth
mentioning that the GDS4206 consistently showed
low efficacy, not just with PathDisGene but across
other G-S-M tools as well, such as (Qumsiyeh,
Jayousi 2021, Qumsiyeh et al., 2022; Qumsiyeh,
Salah, et al., 2023; Yousef et al., 2019).
Datasets GDS2545 and GDS2547 exhibit
moderate performance, with AUC values of 74.9%
and 73.6%, respectively, alongside adequately
balanced sensitivity and specificity measures. These
results underscore their moderate discriminatory
skills relative to other datasets in the table.
Table 3 highlights the variability in
PathDisGene's efficacy across several datasets,
notably influenced by the quantity and quality of
genes linked to each dataset. High-performing
datasets like GDS3837 and GDS1962 illustrate the
model's capabilities while lower-performing datasets
like GDS4206 underscore the difficulties of
employing generalized methodologies on datasets
with distinct attributes.
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
680
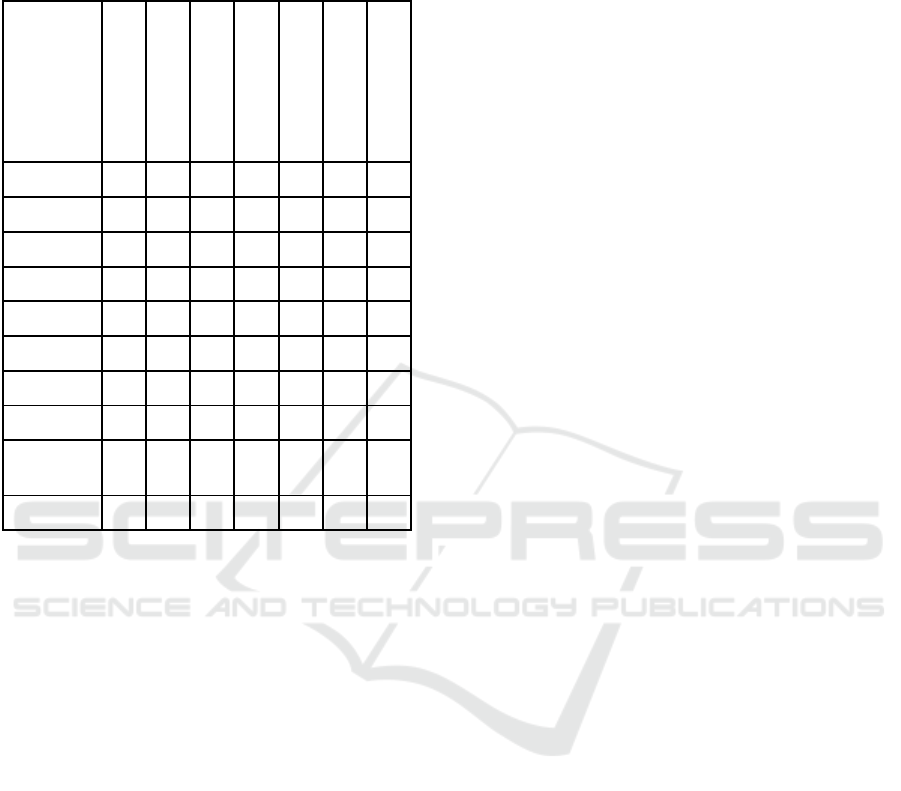
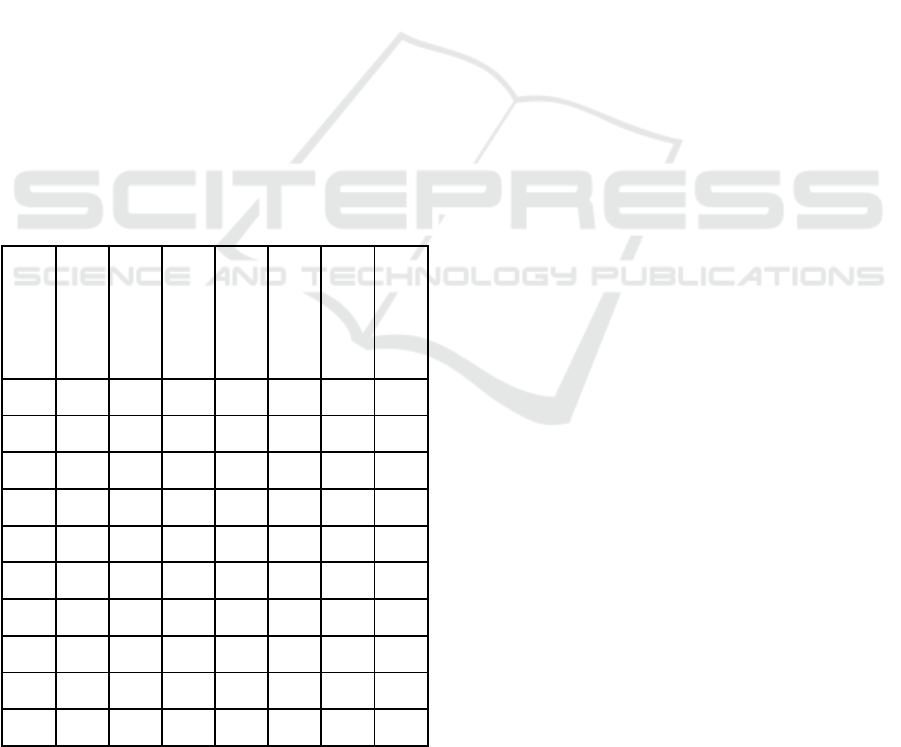
Table 3: An Overview of PathDisGene Performance
Metrics. This table presents the Accuracy, Sensitivity,
Specificity, Precision, and F-measure for 10 GEO datasets
for the top two ranked groups.
GEO accession
# of Features
Sensitivity
Specificity
Precision
Accuracy
Area Under Curve
F-measure
GDS1962 3.11 0.91 0.81 0.93 0.88 0.93 0.91
GDS2545 6 0.66 0.70 0.73 0.68 0.74 0.68
GDS2547 4.45 0.68 0.67 0.68 0.67 0.73 0.67
GDS2771 3.9 0.62 0.64 0.66 0.63 0.67 0.63
GDS3257 3.9 0.62 0.64 0.66 0.63 0.67 0.63
GDS3268 3.55 0.54 0.56 0.58 0.55 0.60 0.56
GDS3837 3.71 0.87 0.90 0.91 0.89 0.94 0.88
GDS4206 2.89 0.36 0.77 0.43 0.64 0.61 0.46
GDS4516
_
4718
2.89 0.36 0.77 0.43 0.64 0.61 0.46
GDS5499 4.13 0.87 0.7 0.87 0.82 0.85 0.87
6 CONCLUSIONS
PathDisGene is a novel machine-learning tool that
represents a notable progression in bioinformatics. It
integrates biological knowledge with machine
learning to tackle the complex nature of pathway-
disease associations. The tool utilizes the G-S-M
approach to efficiently categorize and prioritize genes
related to specific disease associations, enhancing
accuracy and stability in disease state predictions
across various datasets. PathDisGene differs from
traditional approaches that exclusively identify
significant genes for computational tasks without
utilizing existing biological knowledge by including
disease-pathway associations to reveal more
profound insights.
The study emphasizes the capability of
PathDisGene to uncover previously unrecognized
biological connections, such as common pathways or
biomarkers across many diseases, which may guide
innovative therapy strategies. PathDisGene enhances
the biological significance of its predictions by
methodically employing prior biological knowledge
from databases such as CTD. Despite its
effectiveness, specific datasets highlight the
difficulties of implementing universal approaches
across varied biological contexts, presenting chances
for enhancement. PathDisGene offers a robust and
scalable methodology for identifying essential
pathway-disease associations facilitating progress in
personalized medicine, systems biology, and disease
research.
REFERENCES
Barrett, T., Wilhite, S. E., Ledoux, P., Evangelista, C., Kim,
I. F., Tomashevsky, M., Marshall, K. A., Phillippy, K.
H., Sherman, P. M., Holko, M., Yefanov, A., Lee, H.,
Zhang, N., Robertson, C. L., Serova, N., Davis, S., &
Soboleva, A. (2013). NCBI GEO: Archive for
functional genomics data sets--update. Nucleic Acids
Research, 41(Database issue), D991-995.
https://doi.org/10.1093/nar/gks1193Smith, J. (1998).
Dalianis, H. (2018). Evaluation Metrics and Evaluation. In
H. Dalianis (Ed.), Clinical Text Mining: Secondary Use
of Electronic Patient Records (pp. 45–53). Springer
International Publishing. https://doi.org/10.1007/978-
3-319-78503-5_6
Davis, A. P., Grondin, C. J., Johnson, R. J., Sciaky, D.,
Wiegers, J., Wiegers, T. C., & Mattingly, C. J. (2021).
Comparative Toxicogenomics Database (CTD):
Update 2021. Nucleic Acids Research, 49(D1), D1138–
D1143. https://doi.org/10.1093/nar/gkaa891
Ersoz, N. S., Bakir-Gungor, B., & Yousef, M. (2023).
GeNetOntology: Identifying affected gene ontology
terms via grouping, scoring, and modeling of gene
expression data utilizing biological knowledge-based
machine learning. Frontiers in Genetics, 14.
https://www.frontiersin.org/articles/10.3389/fgene.202
3.1139082
Hsu, S.-D., Lin, F.-M., Wu, W.-Y., Liang, C., Huang, W.-
C., Chan, W.-L., Tsai, W.-T., Chen, G.-Z., Lee, C.-J.,
Chiu, C.-M., Chien, C.-H., Wu, M.-C., Huang, C.-Y.,
Tsou, A.-P., & Huang, H.-D. (2011). miRTarBase: A
database curates experimentally validated microRNA–
target interactions. Nucleic Acids Research,
39(suppl_1), D163–D169. https://doi.org/10.1093/nar/
gkq1107
Jabeer, A., Temiz, M., Bakir-Gungor, B., & Yousef, M.
(2023). miRdisNET: Discovering microRNA
biomarkers that are associated with diseases utilizing
biological knowledge-based machine learning.
Frontiers in Genetics, 13. https://www.frontiersin.org/
articles/10.3389/fgene.2022.1076554
Jin, L., Zuo, X.-Y., Su, W.-Y., Zhao, X.-L., Yuan, M.-Q.,
Han, L.-Z., Zhao, X., Chen, Y.-D., & Rao, S.-Q. (2014).
Pathway-Based Analysis Tools for Complex Diseases:
A Review. Genomics, Proteomics & Bioinformatics,
12(5), 210–220. https://doi.org/10.1016/j.gpb.2014.10.
002
Łukasiewicz, S., Czeczelewski, M., Forma, A., Baj, J.,
Sitarz, R., & Stanisławek, A. (2021). Breast Cancer—
Epidemiology, Risk Factors, Classification, Prognostic
PathDisGene: Discovering Informative Gene Groups for Disease Diagnosis Using Pathway-Disease Associations and a Grouping, Scoring,
Modeling-Based Machine Learning Approach
681

Markers, and Current Treatment Strategies—An
Updated Review. Cancers, 13(17), 4287.
https://doi.org/10.3390/cancers13174287
MacEachern, S. J., & Forkert, N. D. (2021). Machine
learning for precision medicine. Genome, 64(4), 416–
425. https://doi.org/10.1139/gen-2020-0131
Qumsiyeh, E., Bakir-Gungor, B., & Yousef, M. (2024).
Classification of Breast Cancer Molecular Subtypes
with Grouping-Scoring-Modeling Approach that
Incorporates Disease-Disease Association Information.
2024 32nd Signal Processing and Communications
Applications Conference (SIU), 1–4. https://doi.org/
10.1109/SIU61531.2024.10601041
Qumsiyeh, E., Salah, Z., & Yousef, M. (2023).
miRGediNET: A comprehensive examination of
common genes in miRNA-Target interactions and
disease associations: Insights from a grouping-scoring-
modeling approach. Heliyon, 9(12), e22666.
https://doi.org/10.1016/j.heliyon.2023.e22666
Qumsiyeh, E., Showe, L., & Yousef, M. (2022). GediNET
for discovering gene associations across diseases using
knowledge based machine learning approach. Scientific
Reports, 12(1), Article 1. https://doi.org/10.1038/s415
98-022-24421-0
Qumsiyeh, E., Yazıcı, M., & Yousef, M. (2023).
GediNETPro: Discovering Patterns of Disease Groups.
Proceedings of the 16th International Joint Conference
on Biomedical Engineering Systems and Technologies
- BIOINFORMATICS, 195–203. https://doi.org/10.52
20/0011690800003414
Qumsiyeh, E., Yousef, M., Salah, Z., & Jayousi, R. (2023).
Detecting Semantic Similarity of Diseases based
Machine Learning. 2023 IEEE International
Conference on Bioinformatics and Biomedicine
(BIBM), 3118–3124. https://doi.org/10.1109/BIBM5
8861.2023.10385728
Qumsiyeh, E., Yousef, M., & Yousef, M. (2024). ReScore
Disease Groups Based on Multiple Machine Learnings
Utilizing the Grouping-Scoring-Modeling Approach:
Proceedings of the 17th International Joint Conference
on Biomedical Engineering Systems and Technologies,
446–453. https://doi.org/10.5220/0012379400003657
Tomczak, K., Czerwińska, P., & Wiznerowicz, M. (2015).
The Cancer Genome Atlas (TCGA): An immeasurable
source of knowledge. Contemporary Oncology, 19(1A),
A68–A77. https://doi.org/10.5114/wo.2014.47136
Xu, Q.-S., & Liang, Y.-Z. (2001). Monte Carlo cross
validation. Chemometrics and Intelligent Laboratory
Systems, 56(1), 1–11. https://doi.org/10.1016/S0169-
7439(00)00122-2
Yousef, M., Abdallah, L., & Allmer, J. (2019). maTE:
Discovering expressed interactions between microRNAs
and their targets. Bioinformatics, 35(20), 4020–4028.
https://doi.org/10.1093/bioinformatics/btz204
Yousef, M., Allmer, J., İnal, Y., & Gungor, B. B. (2024).
G-S-M: A Comprehensive Framework for Integrative
Feature Selection in Omics Data Analysis and Beyond
(p. 2024.03.30.585514). bioRxiv. https://doi.org/
10.1101/2024.03.30.585514
Yousef, M., Bakir-Gungor, B., Jabeer, A., Goy, G.,
Qureshi, R., & C. Showe, L. (2021). Recursive Cluster
Elimination based Rank Function (SVM-RCE-R)
implemented in KNIME. F1000Research, 9, 1255.
https://doi.org/10.12688/f1000research.26880.2
Yousef, M., Goy, G., & Bakir-Gungor, B. (2022).
miRModuleNet: Detecting miRNA-mRNA Regulatory
Modules. Frontiers in Genetics, 13, 767455.
https://doi.org/10.3389/fgene.2022.767455
Yousef, M., Goy, G., Mitra, R., Eischen, C. M., Jabeer, A.,
& Bakir-Gungor, B. (2021). miRcorrNet: Machine
learning-based integration of miRNA and mRNA
expression profiles, combined with feature grouping
and ranking. PeerJ, 9, e11458. https://doi.org/10.7717/
peerj.11458
Yousef, M., Jabeer, A., & Bakir-Gungor, B. (2021). SVM-
RCE-R-OPT: Optimization of Scoring Function for
SVM-RCE-R. In G. Kotsis, A. M. Tjoa, I. Khalil, B.
Moser, A. Mashkoor, J. Sametinger, A. Fensel, J.
Martinez-Gil, L. Fischer, G. Czech, F. Sobieczky, & S.
Khan (Eds.), Database and Expert Systems
Applications—DEXA 2021 Workshops (pp. 215–224).
Springer International Publishing. https://doi.org/10.10
07/978-3-030-87101-7_21
Yousef, M., Ozdemir, F., Jaaber, A., Allmer, J., & Bakir-
Gungor, B. (2022). PriPath: Identifying Dysregulated
Pathways from Differential Gene Expression via
Grouping, Scoring and Modeling with an Embedded
Machine Learning Approach [Preprint]. In Review.
https://doi.org/10.21203/rs.3.rs-1449467/v1
Yousef, M., Ülgen, E., & Uğur Sezerman, O. (2021).
CogNet: Classification of gene expression data based
on ranked active-subnetwork-oriented KEGG pathway
enrichment analysis. PeerJ. Computer Science, 7, e336.
https://doi.org/10.7717/peerj-cs.336
Dalianis, H. (2018). Evaluation Metrics and Evaluation. In
H. Dalianis (Ed.), Clinical Text Mining: Secondary Use
of Electronic Patient Records (pp. 45–53). Springer
International Publishing. https://doi.org/10.1007/978-
3-319-78503-5_6
Davis, A. P., Grondin, C. J., Johnson, R. J., Sciaky, D.,
Wiegers, J., Wiegers, T. C., & Mattingly, C. J. (2021).
Comparative Toxicogenomics Database (CTD):
Update 2021. Nucleic Acids Research, 49(D1), D1138–
D1143. https://doi.org/10.1093/nar/gkaa891
Hsu, S.-D., Lin, F.-M., Wu, W.-Y., Liang, C., Huang, W.-
C., Chan, W.-L., Tsai, W.-T., Chen, G.-Z., Lee, C.-J.,
Chiu, C.-M., Chien, C.-H., Wu, M.-C., Huang, C.-Y.,
Tsou, A.-P., & Huang, H.-D. (2011). miRTarBase: A
database curates experimentally validated microRNA–
target interactions. Nucleic Acids Research,
39(suppl_1), D163–D169. https://doi.org/10.1093/nar/
gkq1107
Jabeer, A., Temiz, M., Bakir-Gungor, B., & Yousef, M.
(2023). miRdisNET: Discovering microRNA
biomarkers that are associated with diseases utilizing
biological knowledge-based machine learning.
Frontiers in Genetics, 13. https://www.frontiersin.org/
articles/10.3389/fgene.2022.1076554
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
682

Jin, L., Zuo, X.-Y., Su, W.-Y., Zhao, X.-L., Yuan, M.-Q.,
Han, L.-Z., Zhao, X., Chen, Y.-D., & Rao, S.-Q. (2014).
Pathway-Based Analysis Tools for Complex Diseases:
A Review. Genomics, Proteomics & Bioinformatics,
12(5), 210–220. https://doi.org/10.1016/j.gpb.2014.10.
002
Łukasiewicz, S., Czeczelewski, M., Forma, A., Baj, J.,
Sitarz, R., & Stanisławek, A. (2021). Breast Cancer—
Epidemiology, Risk Factors, Classification, Prognostic
Markers, and Current Treatment Strategies—An
Updated Review. Cancers, 13(17), 4287.
https://doi.org/10.3390/cancers13174287
MacEachern, S. J., & Forkert, N. D. (2021). Machine
learning for precision medicine. Genome, 64(4), 416–
425. https://doi.org/10.1139/gen-2020-0131
Tomczak, K., Czerwińska, P., & Wiznerowicz, M. (2015).
The Cancer Genome Atlas (TCGA): An immeasurable
source of knowledge. Contemporary Oncology,
19(1A), A68–A77. https://doi.org/10.5114/wo.2014.47
136
Xu, Q.-S., & Liang, Y.-Z. (2001). Monte Carlo cross
validation. Chemometrics and Intelligent Laboratory
Systems, 56(1), 1–11. https://doi.org/10.1016/S0169-
7439(00)00122-2
Yousef, M., Allmer, J., İnal, Y., & Gungor, B. B. (2024).
G-S-M: A Comprehensive Framework for Integrative
Feature Selection in Omics Data Analysis and Beyond
(p. 2024.03.30.585514). bioRxiv. https://doi.org/10.11
01/2024.03.30.585514
Qumsiyeh, E., & Jayousi, R. (2021, November).
Biomedical information extraction pipeline to identify
disease-gene interactions from PubMed breast cancer
literature. In 2021 International Conference on
Promising Electronic Technologies (ICPET) (pp. 1-6).
IEEE.
Qumsiyeh, E (2024). Enhancing Breast Cancer Subtype
Classification through GediNET: Integrating Disease-
Disease Association Data with a Grouping-Scoring-
Modeling Approach.
Holzinger, A., Goebel, R., Palade, V., & Ferri, M. (2017).
Towards integrative machine learning and knowledge
extraction. In Towards Integrative Machine Learning
and Knowledge Extraction: BIRS Workshop, Banff, AB,
Canada, July 24-26, 2015, Revised Selected Papers
(pp. 1-12). Springer International Publishing.
PathDisGene: Discovering Informative Gene Groups for Disease Diagnosis Using Pathway-Disease Associations and a Grouping, Scoring,
Modeling-Based Machine Learning Approach
683
