
Towards a New Method for Perturbation Analysis in Biochemical
Pathways Based on Network Propagation
Niccol
`
o De Paolis and Paolo Milazzo
a
Department of Computer Science, Univesity of Pisa, Largo B. Pontecorvo 3, Pisa, Italy
Keywords:
Network Propagation, Biological Networks, Pathways, Chemical Kinetics.
Abstract:
We introduce a preliminary definition of a network propagation approach to tackle the problem of investigating
the spread of mutation-induced perturbations in biochemical pathways relying on network topology alone,
without the need for quantitative details such as species concentrations and kinetic constants of reactions
required to model the trajectory of species concentrations using stochastic and deterministic algorithms. These
details are not always available, hence our goal is to provide insights regarding the impact of perturbations even
when lacking such information. We further describe the definition of a synthetic dataset the algorithm has been
tested on and provide the results obtained in terms of accuracy in identifying the effect of the perturbation on
each species. Finally, a real world scenario is presented in order to show the potential of the proposed solution
and spot its possible limitations.
1 INTRODUCTION
Systems biology (Kitano, 2002) deals with the def-
inition and analysis of biological processes, defined
as systems made by interacting entities. When con-
sidering the systems at a molecular level, they can be
seen as networks of chemical reactions. The analy-
sis of the dynamics of such systems is particularly in-
teresting to better elucidate how biological processes
are carried on and how sensitive they are to pertur-
bations such as changes of species concentrations or
of kinetic parameters of chemical reactions. Chemi-
cal reaction networks (in particular, biochemical path-
ways) can be analyzed by means of simulations meth-
ods. The two most common approaches are the deter-
ministic and the stochastic ones. The former makes
use of systems of Ordinary Differential Equations
(ODEs) to describe the trajectory of the concentration
of each species over time in a continuous way. The
latter is based on algorithms such as Gillespie’s SSA
(Gillespie, 2007) to model the evolution of concen-
trations with discrete quantities and at discrete time
steps. Both approaches can provide very accurate re-
sults, but they can become computationally demand-
ing, in particular in the case of stiff systems, of highly
stochastic systems and when several parameter con-
figurations have to be tested. Furthermore, simula-
a
https://orcid.org/0000-0002-7309-6424
tion approaches suffer from a major drawback, which
is the need for a complete and detailed description of
the system under consideration, where detailed means
that they require the knowledge of initial concentra-
tions of all species and of kinetic constants of all re-
actions.
Unfortunately, concentration values and kinetic
parameters of biochemical reaction networks are of-
ten not available. In order to overcome this limi-
tation, different approaches requiring a less detailed
description emerged. These methods are based on
the hypothesis that the structural properties of a path-
way correlate with its dynamical properties (Bailey,
2001). Several methods have been proposed to in-
fer knowledge on the pathway dynamics from the
structure of its graph representation without perform-
ing time-consuming simulations. In (Craciun et al.,
2006; Angeli et al., 2010; Yordanov et al., 2020), ap-
proaches based on chemical reaction network theory
are proposed and implemented in order to detect spe-
cific dynamical properties such as multi-stability or
robustness. Other approaches based on the same hy-
potheses exploit machine learning methods for graphs
to infer dynamical properties from topological ones
(Fontanesi et al., 2023; Bove et al., 2020).
Many questions can be asked regarding the dy-
namics of biochemical systems. In this work, we aim
to investigate how the impact of perturbations of ki-
netic constants spread in biochemical pathways, influ-
684
De Paolis, N. and Milazzo, P.
Towards a New Method for Perturbation Analysis in Biochemical Pathways Based on Network Propagation.
DOI: 10.5220/0013384200003911
Paper published under CC license (CC BY-NC-ND 4.0)
In Proceedings of the 18th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2025) - Volume 1, pages 684-691
ISBN: 978-989-758-731-3; ISSN: 2184-4305
Proceedings Copyright © 2025 by SCITEPRESS – Science and Technology Publications, Lda.

encing the concentration of each species involved in
the pathway.
A perturbation of a kinetic constant in a pathway
could be due to several causes. For instance, a muta-
tion of a protein could increase (or decrease) its bind-
ing affinity for a given ligand, making the reaction de-
scribing the binding more (or less) likely. This can be
captured by an increased (or decreased) kinetic con-
stant. Similarly, a specific environmental condition
could favor (or disfavor) a specific chemical reaction,
and this again could be captured by a perturbation in
its kinetic constant. In these cases, the dynamics of
the perturbed chemical reaction changes, causing a
change in the concentrations of its reactants and prod-
ucts which, in turn, could be involved in other reac-
tions. This causes also the other reactions to change
their dynamics, triggering a propagation process.
A similar problem has been tackled in the litera-
ture, where the perturbation considered is at the level
of species concentrations rather than chemical reac-
tions’ kinetics (Santolini and Barab
´
asi, 2018). In the
aforementioned study, the problem is tackled using a
network propagation approach and the goal is to as-
sess whether a given species received a positive or
negative impact from the alteration. The accuracy ob-
tained in this study is 65%, with an increase up to 80%
in pathways satisfying specific network properties. In
the same study, some characteristics of the biochemi-
cal pathways are presented as problematic (i.e., pres-
ence of reversible reactions and very fast reactions).
The approach we propose in this paper shares the
goals of the one in (Santolini and Barab
´
asi, 2018): un-
derstanding the effects of a perturbation in a pathway
from a global point of view by relying on topology
alone, so without the access to often unavailable infor-
mation such as kinetic constants. Our propagation ap-
proach is based on an algorithm that is inspired by the
chemical reaction theory, and mimics in an abstract
domain the computation of reaction rates and the ef-
fects that reactions have on their reactants and prod-
ucts concentrations. This will allow our method to
perform better than the one in (Santolini and Barab
´
asi,
2018) in the classes of pathways that in such a pa-
per are identified as particularly problematic. Indeed,
we will show that on a dataset of synthetic pathways
designed to focus on the problematic topological fea-
tures, our algorithm exhibits an accuracy of 83.95%.
The development of our approach is still ongoing,
but the preliminary results we present in this paper
suggest that, with further development, it could be
successfully applied to real-world pathways.
This paper is structured as follows. In Section 2
we provide a brief description of biochemical path-
ways as networks of chemical reactions and introduce
the computational tools and data types commonly
used to perform analysis of their dynamics. In Sec-
tion 3 we provide the intuitions behind the develop-
ment of a network propagation algorithm tailored for
chemical reaction networks. In Section 4 we describe
the process that lead to the definition of a dataset of
synthetic pathways the algorithm has been first tested
on. In Section 5 we describe the experimental setup
and some results obtained on the synthetic dataset. In
Section 6 we present a case study in which the propa-
gation algorithm is applied to a real pathway. Finally,
in Section 7 we draw some conclusions and highlight
future directions for this work.
2 BACKGROUND
As already stated, biological systems can be repre-
sented from a molecular point of view as chemical
reaction networks. A single chemical reaction can be
seen as a process that converts some input compounds
into output ones. Formally:
l
1
R
1
+ l
2
R
2
+ ... + l
i
R
i
k
−→ l
′
1
P
1
+ l
′
2
P
2
+ ... + l
′
j
P
j
where the l and l
′
values represent the stoichiomet-
ric coefficients, instances of R and P represent the
molecules taking part in the reaction (reactants and
products, respectively) and k ∈ R
>0
is the kinetic con-
stant of the reaction. Reactants concentrations and the
kinetic constant k serve to compute another quantity
called reaction rate, which defines how frequently the
reaction takes place (i.e., how frequently reactants are
decreased and products are increased). According to
the mass-action law, the reaction rate is proportional
to the availability of reactants and to the kinetic con-
stant of the reaction. Hence, by denoting the concen-
tration of a reactant R as [R], the reaction rate is math-
ematically defined as
r = k[R
1
]
l
1
···[R
i
]
l
i
.
Given this definition and the fact every time a reac-
tion takes place the concentration of both reactants
and products changes, it is clear that a change in ei-
ther the reactant concentrations or the kinetic constant
will impact the trajectory of concentrations of both re-
actants and products.
Given a pathway expressed as a set of chemical re-
actions, the dynamics of species concentrations over
time can be analyzed by using the reaction rates to
construct a system of Ordinary Differential Equations
(ODEs). The system will contain one variable and one
equation for every molecular species, and the equa-
tion will be essentially given by the summation of
Towards a New Method for Perturbation Analysis in Biochemical Pathways Based on Network Propagation
685

rates of the reactions in which such a molecule is in-
volved, either as reactant (with negative sign in the
equation) or as product (with positive sign). Such a
system of ODEs can then be analyzed by applying a
standard numerical integration method (e.g., a method
of the Runge-Kutta family). Alternatively, reaction
rates defined in a slightly different way could be
used to simulate the dynamics of concentrations us-
ing a stochastic simulation method such as Gillespie’s
stochastic simulation algorithm (SSA). For more de-
tails on both deterministic (ODE-based) and stochas-
tic analysis approaches, we refer the reader to (Gratie
et al., 2013; Gillespie, 2007).
From a computational point of view, biochemical
pathways can be represented using the SBML file for-
mat (Hucka et al., 2003). This format is general and
application-agnostic, in the sense that it serves as a
way to just model reactions-based systems. Such a
representation can then be analyzed in many differ-
ent ways using many different tools, thus reusing the
same definition for a wide range of purposes. Sev-
eral repositories of pathways are available nowadays,
such as KEGG (Kanehisa, 2002), Reactome (Milacic
et al., 2024), and BioModels (Malik-Sheriff et al.,
2020). All of them include the possibility of down-
lowding SBML representations of pathways. How-
ever, among these, BioModels (Malik-Sheriff et al.,
2020) is the one more focused on pathways ready for
simulation, namely which include kinetic constants,
concentrations, and so on. BioModels contains over
one thousand manually curated SBML files. Some-
one wishing to create or interact with this file for-
mat can leverage, for example, the LibSBML library
(Bornstein et al., 2008) that provides APIs accessi-
ble from Python and other languages. To investigate
dynamical properties of pathways, several libraries
and tool implementing different computational meth-
ods are available, and work on SBML representations
of pathways. One such libraries is LibRoadRunner
(Welsh et al., 2022), that provides an interface to in-
teract with SBML files and also to perform fast simu-
lations in order to understand the evolution in species
concentrations. The library provides the user an easy-
to-use interface to perform many kinds of analysis of
dynamical systems, among which simulation, which
can be carried on either by means both ODE-based
and stochastic methods.
Using the methods and tools we described so far,
the global (pathway-wide) effect of the perturabion of
a kinetic constants could be investigated as follows:
1. Take the SBML representation of the pathaway of
interest, including all kinetic constants and initial
concentration values
2. Run a simulation using a method based on numer-
ical integration of ODEs, or a number of simula-
tions using a stochastic simulation algorithm
3. Repeat the simulation(s) by varying the value of
the perturbed kinetic constant in the SBML repre-
sentation
4. Compare the final concentration of each molecu-
lar species of the pathways obtained from the per-
turbed simulation(s) with those obtained from the
simulation(s) of the original SBML model
5. For each molecular species, classify it as in-
creased or decreased (or, possibly, unchanged)
according to whether the perturbation caused its
final concentration to increase or decrease (or not
to change)
In this paper, we will use this approach based on
simulations in order to construct the ground-truth for
a benchmark set of perturbed pathways. However,
since the use of this method can be hampered by the
unavailability of kinetic constants and initial concen-
tration values, or could be made unfeasible by the
computational cost of running simulations, in Section
3 we will propose an alternative approach based on
network propagation. We will evaluate the quality of
the new approach with respect to the simulation-based
ground truth.
3 A NETWORK PROPAGATION
APPROACH
The approach we propose takes inspiration from
the computational framework of network propagation
(Cowen et al., 2017), which comprises a class of al-
gorithms with the same underlying rationale: the dif-
fusion of information across a network relies on its
topology. Network propagation approaches are of
many kinds and find several applications in the field of
systems biology (Picart-Armada et al., 2019; Charmpi
et al., 2021; Carlin et al., 2017).
In our approach, the source of the propagation is
the reaction whose kinetic constant is perturbed, and
we propagate information about how concentrations
of molecular species are influenced by such a pertur-
bation. However, the information being propagated
in our approach is not the absolute value of the con-
centrations, since we wish to our method to work also
when such information is missing. We instead prop-
agate an abstract value indicating whether a given
species received a positive or negative impact from
the perturbation.
To better elucidate the idea behind the algorithm,
it is first mandatory to describe how a chemical reac-
tion can be represented as a directed bipartite graph,
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
686
namely a graph with two disjoint sets of nodes (one
for the species and one for the reactions) with di-
rected edges only connecting nodes belonging to a set
to nodes belonging to the other. On top of this formu-
lation, one can embed knowledge about the reactions
at different levels of detail. Our algorithm works on
an instance of the bipartite graph (called from now on
pathway graph) that labels each node with one of the
following two quantities that regulate the dynamics:
the species shift and the reaction alteration.
The species shift is a value in [0, 1] representing
in an abstract way how much the final species con-
centrations in the perturbed pathway differ from the
final concentrations in the original pathway. Such val-
ues, used as species node labels, are initially set to 0.5
(representing no difference between original and per-
turbed pathways). The propagation process can lead
each of them to decrease: a value close to 0 means
that the final concentration of the species in the per-
turbed pathway is significantly decreased with respect
to the original pathway. Otherwise, each of the shift
values can increase: a value close to 1 represents a
significant increase in the perturbed pathway.
The reaction alteration is a real value representing
in an abstract way the rate increase (if positive) or de-
crease (if negative) of each reaction in the perturbed
pathway with respect to the original pathway. This
can be directly due to the perturbation or to the effect
of propagation. It is initially set to a non zero value for
the perturbed reaction (we used 2 or −2 in our prelim-
inary experiments) and to zero for the other reactions.
Then the propagation algorithm will lead these values
to change.
The labels embedded in the nodes (shift for
species node and alteration for reaction nodes) are
iteratively updated and are used to discriminate be-
tween positively impacted species and negatively im-
pacted ones. The update functions applied at each
iteration and the termination conditions will be ex-
plained below.
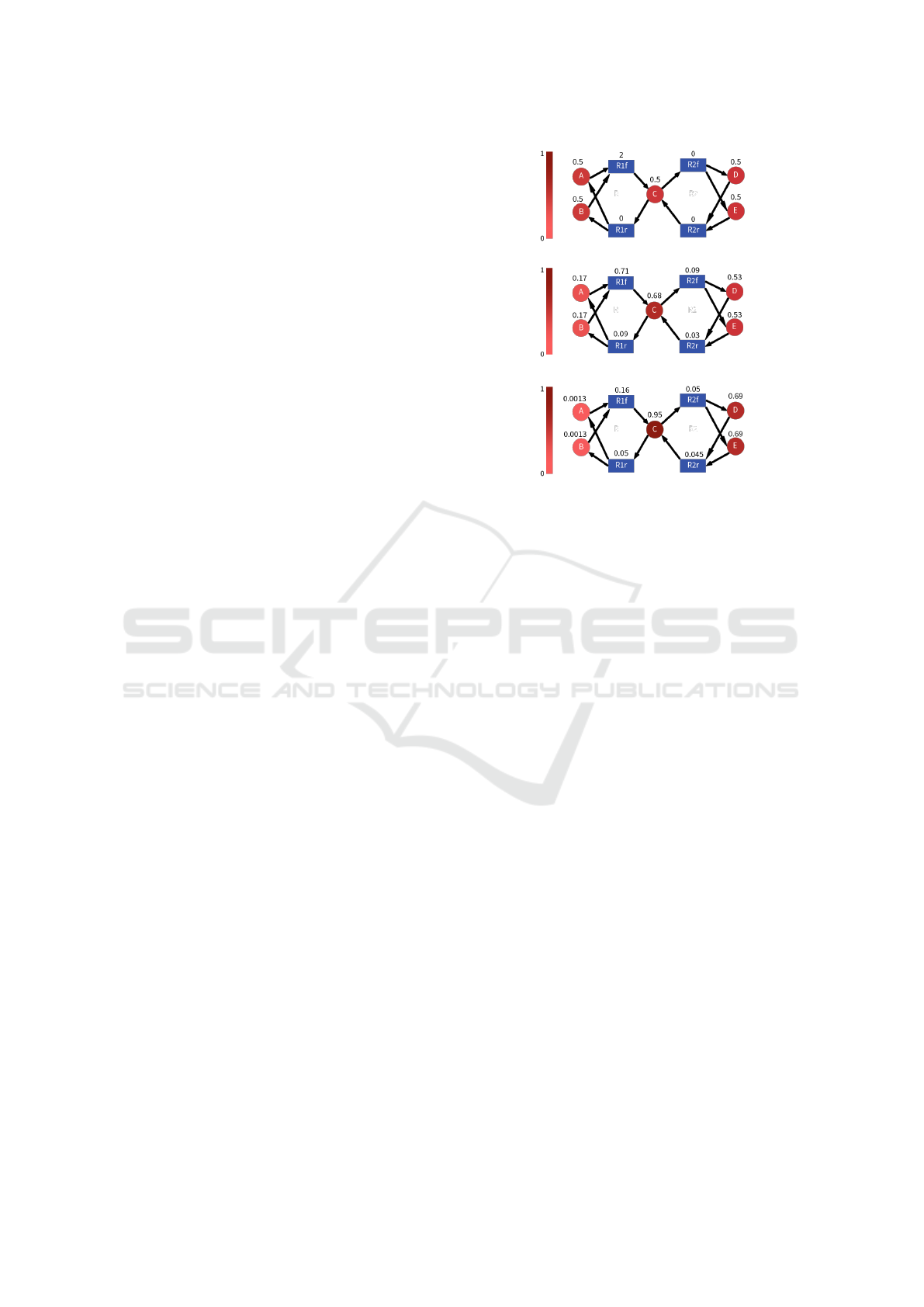
An example of execution of the algorithm is de-
picted in Fig.1, where three snapshots of the node la-
belling are presented corresponding to the initial one
(Fig. 1a), one obtained in the middle of the execution
of the propagation algorithm (Fig. 1b), and the final
one in (Fig. 1c). In this example, we have four reac-
tions (R1 f , R1r, R2 f and R2r, which can also be seen
as two reversible reactions) acting on five species (A,
B, C, D and E). Species A and B are the reactants
of R1 f while C is its product, and so on. As shown in
Fig. 1a, all species shifts are initially set to 0.5 and the
only reaction with non-zero alteration is R1 f , which
is associated to a value 2 representing a perturbation
which increases its rate (corresponding to an increase
(a) Initial labeling.
(b) Intermediate labeling.
(c) Final labeling.
Figure 1: Example of execution of the propagation algo-
rithm.
in its kinetic constant). The algorithm starts iterating:
the positive alteration of R1 f causes the shifts of its
reactants A and B to decrease and the shifts of its prod-
uct C to increase. This, in turn, induces an increase in
the alteration of reactions having C as reactant (since
higher concentration of reactants correspond to higher
reaction rate) and a decrease in the alteration of reac-
tions having A and B as reactants (for the opposite
reason). After a few iterations, the algorithm causes
the node labels to reach the values depicted in Fig.
1b. At the end of the propagation process, node la-
bels reach the values shown in Fig. 1c, representing
the fact that the final concentrations of A and B have
been significantly decreased (shift close to 0) by the
perturbation of R1 f , the final concentration of C has
been significantly increased (shift close to 1), and also
D and E have been increased (shift higher than 0.5).
Species shift and reaction alterations serve to
compute a third quantity: the reaction potential,
which is the value really governing the dynamics. The
potential has been defined in a way to mimic the role
of the reaction rate that regulates how compounds are
consumed and produced in kinetic theory of chemical
reactions. Given a reaction R defined as: A + B
k
−→ C
we can compute the potential for R as
R.potential = ∆A + ∆B + R.alteration
where ∆A and ∆B are the marginal shifts for species
A and B, respectively, namely ∆A = A.shift −0.5 and
∆B = B.shift −0.5. Consequently, the potential is di-
rectly proportional to the reactants shifts, like in ki-
netic theory the reaction rate is proportional to the re-
Towards a New Method for Perturbation Analysis in Biochemical Pathways Based on Network Propagation
687

actants’ concentration. The potential is also propor-
tional to the reaction alteration, like the reaction rate
is proportional to the kinetic constant of the reaction.
As already said, the potential is the value governing
the dynamics. Species values are iteratively updated
by firing reactions, which can be seen as the execu-
tor of an exchange of flow from reactants to prod-
ucts. The flow conveys an information which is pro-
portional to the state of reactants (the shift) and to the
state of the reactions (the alteration mark) and vice
versa. Given the reaction introduced above, we can
see how the values are updated according to the fol-
lowing rule:
A.shift = A.shift −R.potential
B.shift = B.shift −R.potential
C.shift = C.shift + R.potential
Defined this way, the values associated to species can
grow or shrink indefinitely. For this reason, their
value is not used as it is, but it first passes though a
squashing function. The function is a sigmoid trans-
lated by 0.5 in order to map the base case of 0.5 still at
0.5. This process restricts the domain in the interval
[0, 1] and a species is seen as increased by the algo-
rithm if its value is in the upper part of the domain
(species.shift ∈ [0.5, 1]), decreased otherwise. A sim-
ilar mechanism drives the computation of the poten-
tial, which is passed through another squashing func-
tion, this time an hyperbolic tangent, in order to re-
strict the domain in [−1, 1] while keeping the sign of
the potential unchanged.
Another mechanism, introduced to force the con-
vergence (otherwise not guaranteed) and to halt the
propagation, is the potential decay. At each iteration,
the potential of each reaction is computed as:
R.potential =
R.potential
√
iteration
This way its value will progressively shrink as the it-
eration count increases, eventually stopping the dy-
namics.
Given this formulation, the algorithm is fully de-
terministic and produces just a single result per net-
work topology. Chemical reaction networks, on the
other hand, may produce different outputs even for the
same network, this is due to the fact that different con-
figurations of kinetic constants may result in different
behaviors. Our algorithm is limited in this sense, as it
fails to capture the possibly larger spectrum of results.
For this reason, we introduced a weight mechanism
to mimic the behavioral variability caused by the dif-
ferent combinations of kinetic constant that could be
present in the original pathway.
Weights are values associated to each reaction that
impact how the potential is computed. In particular,
the weight of a reaction R is a multiplicative constant
w
R
used in the computation of the reaction potential
as follows:
R.potential = w
R
(∆A + ∆B + R.alteration)
Weights are associated to each reaction of the path-
way under study, and in our preliminary experiments
we set them by performing a grid search of size 1000,
drawing each time a uniformly distributed random
value in the interval [−0.2, 0.2]. So, for each path-
way, 1000 executions of the algorithm are run, each
with a different configuration of weights. This gives
rise to not just one final value for each species, but to
1000 different ones. To assign a unique final value to
the shift of each species, we take the median of the
1000 final results obtained.
4 SYNTHETIC PATHWAYS
DATASET
In order to perform a preliminary evaluation of the
proposed method, the approach has been tested on a
dataset of synthetic pathways. Pathways in the dataset
presents three main characteristics: presence of re-
versible reactions, presence of very fast reactions and
no synthesis or degradation reactions. These features
were chosen in order to test our approach on scenar-
ios that have been identified as particularly difficult to
tackle in previous approaches available in the litera-
ture (Santolini and Barab
´
asi, 2018).
The dataset of synthetic pathways has been gen-
erated by first performing an analysis of real path-
ways from the BioModels database. The analysis fo-
cused first on the number of reactions and species
that real pathways contain. After this investigation,
it came out that the average number of species taking
part in the biochemical pathways stored in BioMod-
els is 22, while the average number of reactions is
29. The distributions are very skewed. In particular,
it has been noticed how 55% of pathways include at
most 10 species, and less than 10% of the pathways is
made by more than 50 species. Similar considerations
also hold for the number of reactions in each pathway,
where about 80% of the entries is below 30 reactions.
A similar analysis has been carried on the indi-
vidual reactions. One key factor that determines the
topology of the pathways and hence of the graphs
generated from them is the in degree and out degree
of reaction nodes (so the number of reactants and
products of each reaction). It has been noticed that
these values are very small, with 99.6% of reactions
having at most 3 reactants and 99.1% having at most
3 products.
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
688
Biochemical pathways dynamics depends not
only on the pathway topology, but also on the param-
eters that govern its behavior (namely the kinetic con-
stants). For this reason, the analysis also focused on
the distribution of these values. We analyzed in par-
ticular the ratio between the forward and reverse ki-
netic constants of reversible reactions, as it indicates
whether they are unbalanced toward one side or the
other of the reaction and this will impact the state
reached at equilibrium. It came out that the distri-
bution of kinetic constants ratios is very broad, with
values that range from infinitesimal ones to very large
numbers.
After all of these considerations, we generated
three classes of pathways in our dataset, grouped ac-
cording to their size: a small class containing path-
ways with at most 5 reversible reactions, a medium
class with pathways of at most 10 reversible reac-
tions and a large one with pathways of at most 30
reversible reactions. The distribution of inDegree and
outDegree has been chosen to reflect the real ones
presented above, so by avoiding hub nodes with a very
high connectivity degree. Also the kinetic constants
of the reactions have been chosen by considering the
observation made above relative to the fact that re-
versible reactions can be (and often are) very unbal-
anced.
In the end, we generated a set of artificial path-
ways topologies, and for each one we instantiated dif-
ferent synthetic pathways, each having different ratios
of the kinetic constants associated to reversible reac-
tions. Then, a set of experiments has been created.
Each experiment consists in the perturbation of just
one kinetic constant, of one of the reactions belong-
ing to the pathway. The perturbation consists in a 100
fold increase in the kinetic constant value.
We simulated the dynamics of all the generated
pathways as well as of all the perturbed variants. Sim-
ulations have been executed by using LibRoadRunner
and a ODE-based method, as described in Sect. 2. For
each species of each pathway, the difference in final
concentration between the perturbed and the original
case was used to determine whether the species was
increased or decreased by the perturbation. In the
next section, we will use this classification as ground-
truth for the evaluation of our propagation method.
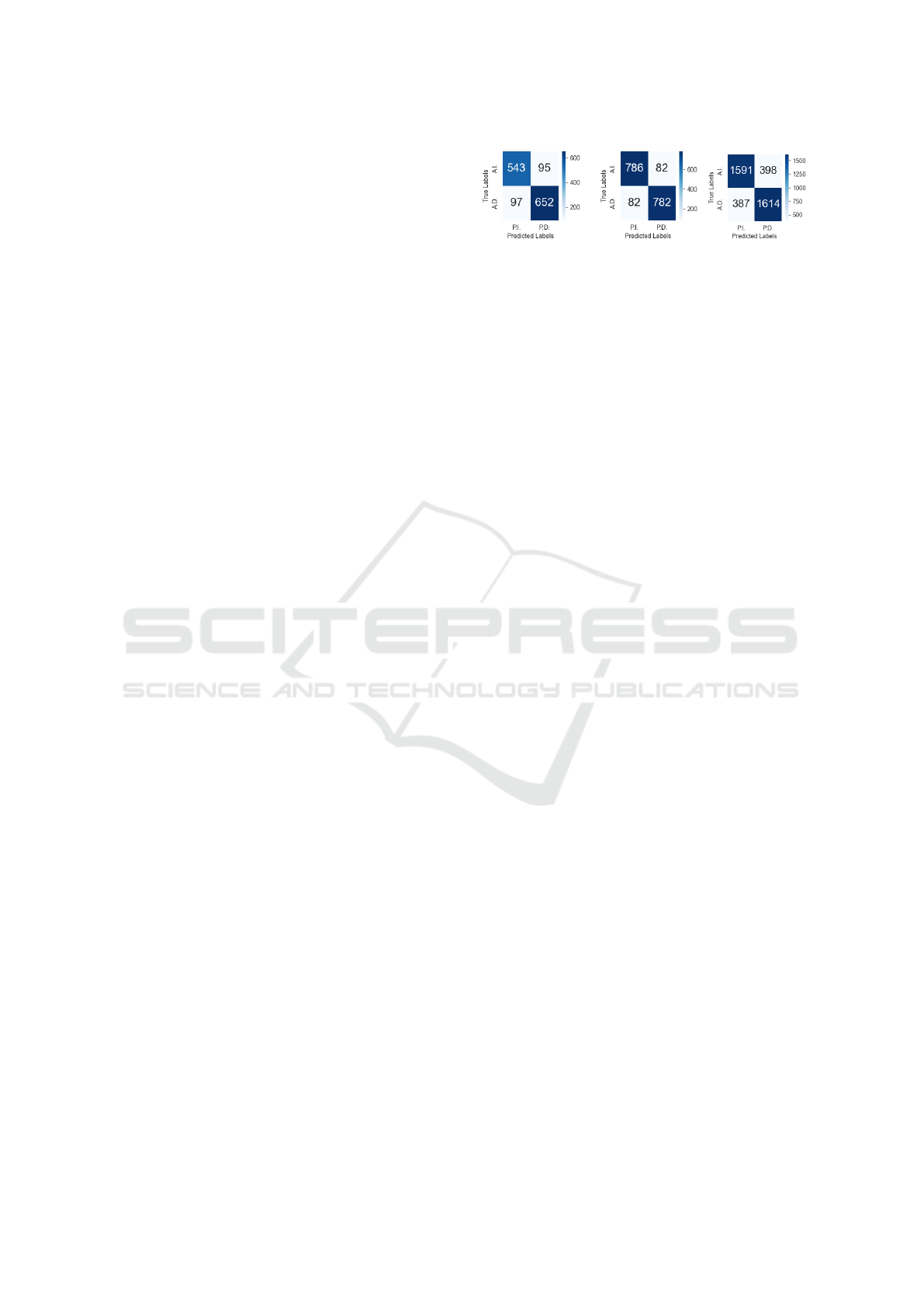
(a) Small class. (b) Medium class. (c) Large class.
Figure 2: Confusion matrices for synthetic dataset. In the
figures, A.I. stands for actual increase, A.D. for actual de-
crease and P.I. and P.D. for predicted increase and predicted
decrease, respectively.
5 EXPERIMENTAL EVALUATION
OF THE PROPAGATION
METHOD
As stated in the previous section, an experiment con-
sists of two pathways: a base, unaltered one, and a
second one where just one reaction has been altered
by increasing its kinetic constant by a factor of 100.
The evaluation phase then consists of two steps: first,
the two pathways are simulated using LibRoadRun-
ner. The species belonging to the pathways are as-
signed a label using the following criterion: the label
is 1 if the concentration of the species in the altered
case is greater than the concentration in the unaltered
one, the label is −1 otherwise. These labels represent
the ground truth values the algorithm’s output will be
compared to. Then our propagation algorithm is run
on the pathway graph associated to the altered path-
way, so the one obtained by marking the affected re-
action with a 2. At the end of the procedure, each
species will be assigned a label following the process
described in Section 3. Using this pipeline, the ap-
proach has been tested on the synthetic dataset and
the results obtained are summarized in Fig.2, where
we can see the confusion matrices for each class of
pathways.
The results we obtained correspond to the follow-
ing accuracies: 86.16% for the small class, 90.53%
for the medium class and 80.33% for the large one.
Overall, the accuracy on the whole dataset is 83, 95%.
These values are higher than those obtained in (San-
tolini and Barab
´
asi, 2018) (65% with an increase up
to 80% in a pathway with specific topological proper-
ties). This comparison is however to be taken just as
a stimulus for the further development of our method.
Indeed, we worked on a synthetic dataset, while the
approach proposed in (Santolini and Barab
´
asi, 2018)
was applied to real pathways. On the other hand, we
focused on the pathways topologies that were identi-
fied as problematic in (Santolini and Barab
´
asi, 2018),
and this makes us confident about the results we could
Towards a New Method for Perturbation Analysis in Biochemical Pathways Based on Network Propagation
689
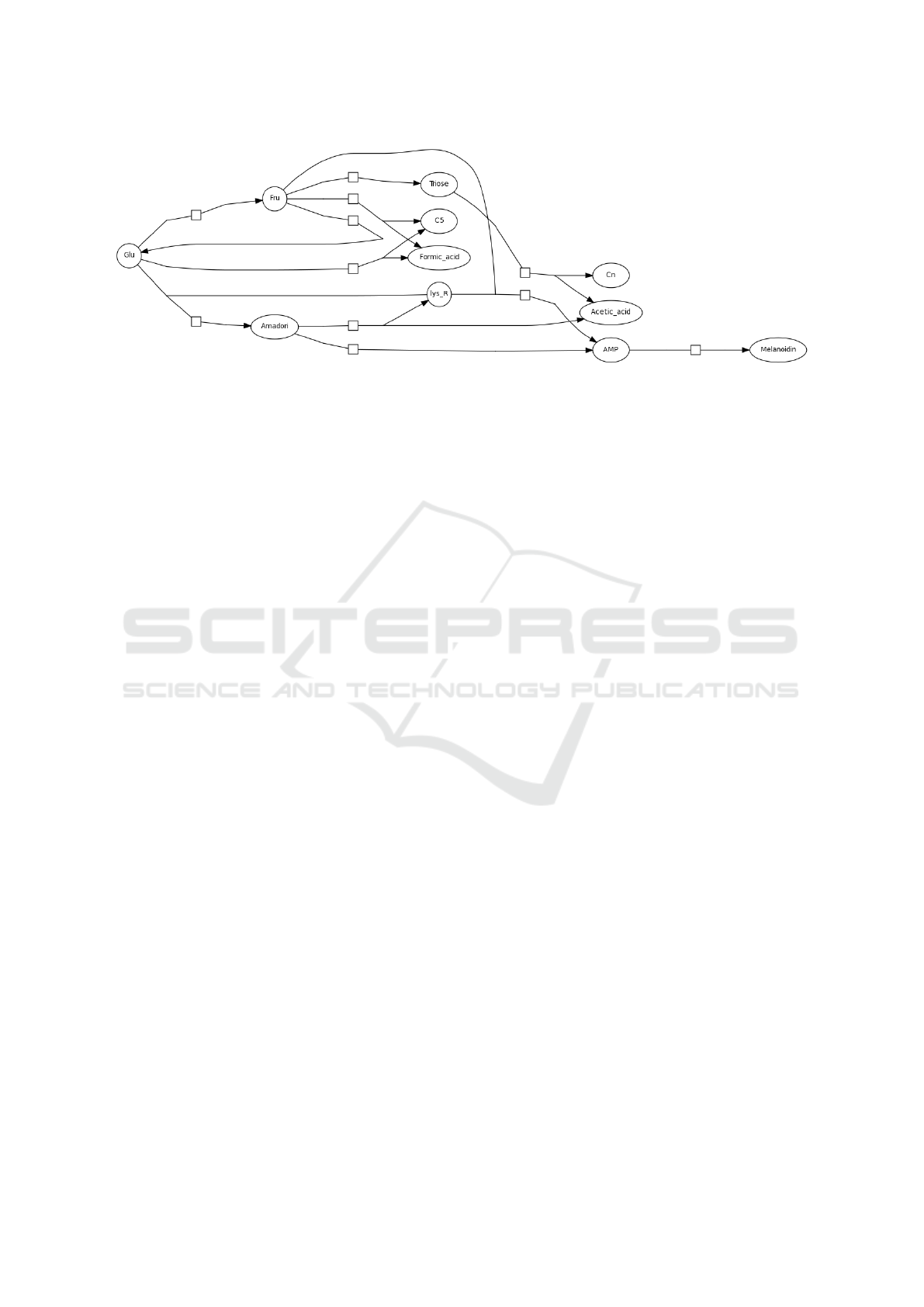
Figure 3: Monosaccharide-casein systems (Pathway BIOMD0000000052 from BioModels) presenting accumulation nodes.
obtain also on real pathways after with further devel-
opments.
6 PROBLEMS TO BE FACED IN
REAL PATHWAYS
Real pathways, as those in BioModels, present some
differences from the synthetic ones we generated. The
main differences are topological, and were clear from
the beginning: the algorithm has been tested and de-
veloped using a benchmark satisfying some strong
assumptions on the network structure (reversible re-
actions, presence of fast reactions and no synthe-
sis/degradation). The motivations for these con-
straints have been already discussed: they were prob-
lematic to be dealt with by previous proposed propa-
gation methods, so we specifically addressed them.
Real pathways, on the other hand, may present
irreversible reactions, synthesis or degradation reac-
tions and also source and sink nodes (species with no
in-edges and out-edges respectively). Our propaga-
tion algorithm can be run even under these topolo-
gies, but we suppose that these topological features
may lead to incorrect results. One of the potential
problem, to give an example, is the fact that in these
cases there could be species that, instead of reaching
a dynamic equilibrium, reach a concentration which
tends to 0 or to a saturation value S. A perturbation
could accelerate or slow down the achievement of the
near 0 or near S concentration, but from the point of
view of the final value reached, the difference could
be negligible. This hampers the work of our pertur-
bation algorithm, which instead is designed in order
to stabilize. However, these are situations that could
be identified quite easily by inspecting the pathway
topology, so we believe that our approach could be
easily extended to take care also of these cases.
To show an example of a real pathway and discuss
the problems arising from the presence of accumula-
tor nodes, we consider the pathway with BioModels
ID BIOMD0000000052 (Fig.3). The application of
our method to this pathway may reveal the impact of
perturbations in one of the reactions in which glucose
(Glu) is involved. For example, an increased trans-
formation into fructose (Fru) may have an impact into
the competing reactions (those in the lower part of the
graph) leading to change in the reached equilibrium.
Such a change could be predicted by our propagation
method. However, this pathway presents many accu-
mulator nodes. An example is Menanoidin (bottom-
right of the pathway) that presents no outgoing edge.
These nodes can easily lead the current version of our
algorithm to incorrect predictions. However, for the
current study we explicitly chose not to consider these
types of nodes, so we are confident to be able to deal
with them as a further development of our method.
7 CONCLUSIONS AND FUTURE
WORK
In this paper, we have presented the ideas behind
the development of a novel network propagation al-
gorithm tailored to work with biochemical pathways
seen as chemical reaction networks. We started by in-
troducing the problem we would like to tackle, that
falls in the domain of dynamics analysis of biochem-
ical pathways, we highlighted the difficulties aris-
ing when approaching the problem using simulation
methods, due to the (very common) unavailability of
kinetic parameters and to the computational cost of
running simulations.
Subsequently, we described the process that led
us to the definition of our propagation algorithm and
also presented a synthetic dataset characterized by
few features identified as problematic in the literature,
and we tested the algorithm on it. Finally, we have
BIOINFORMATICS 2025 - 16th International Conference on Bioinformatics Models, Methods and Algorithms
690

discussed potential problems that our method could
encounter in real world scenarios.
Our algorithm performed very well on our syn-
thetic pathways dataset. The dataset was constructed
to focus on pathway with topologies which have been
previously identified as problematic. This makes us
confident that by proceeding with the development of
our method (e.g., by integrating it with solutions that
have been already explored in the literature) we can be
able to obtain very good results also on real pathways.
Having a method that provides qualitative insights re-
garding the effects of a local perturbations on a global
level would be beneficial to understand, for example,
the alterations caused by a disease-induced mutation
or by a treatment even in cases where current methods
fail due to lack in information. Such method would
be also useful to seamlessly spot configurations that
result in major alterations so that they can be further
studied more accurately.
REFERENCES
Angeli, D. et al. (2010). Graph-theoretic characterizations
of monotonicity of chemical networks in reaction co-
ordinates. J Math Biol, 61:581–616.
Bailey, J. E. (2001). Complex biology with no parameters.
Nature biotechnology, 19(6):503–504.
Bornstein, B. J., Keating, S. M., Jouraku, A., and Hucka, M.
(2008). LibSBML: An API library for SBML. Bioin-
formatics, 24(6):880–881.
Bove, P., Micheli, A., Milazzo, P., and Podda, M.
(2020). Prediction of dynamical properties of bio-
chemical pathways with graph neural networks. In
Proc. of BIOSTEC/Bioinformatics 2020, pages 32–43.
Scitepress.
Carlin, D. E., Demchak, B., Pratt, D., Sage, E., and Ideker,
T. (2017). Network propagation in the cytoscape
cyberinfrastructure. PLoS Computational Biology,
13(10):e1005598.
Charmpi, K., Chokkalingam, M., Johnen, R., and Beyer, A.
(2021). Optimizing network propagation for multi-
omics data integration. PLoS Computational Biology,
17(11):e1009161.
Cowen, L., Ideker, T., Raphael, B. J., and Sharan, R. (2017).
Network propagation: a universal amplifier of genetic
associations. Nature Reviews Genetics, 18(9):551–
562.
Craciun, G. et al. (2006). Understanding bistability in
complex enzyme-driven reaction networks. PNAS,
103:8697–8702.
Fontanesi, M., Micheli, A., Milazzo, P., and Podda, M.
(2023). Exploiting the structure of biochemical path-
ways to investigate dynamical properties with neural
networks for graphs. Bioinformatics, 39(11):btad678.
Gillespie, D. T. (2007). Stochastic simulation of chemical
kinetics. Annu. Rev. Phys. Chem., 58(1):35–55.
Gratie, D.-E., Iancu, B., and Petre, I. (2013). Ode analysis
of biological systems. Formal Methods for Dynamical
Systems: 13th International School on Formal Meth-
ods for the Design of Computer, Communication, and
Software Systems, SFM 2013, Bertinoro, Italy, June
17-22, 2013. Advanced Lectures, pages 29–62.
Hucka, M., Finney, A., Sauro, H. M., Bolouri, H., Doyle,
J. C., Kitano, H., Arkin, A. P., Bornstein, B. J., Bray,
D., Cornish-Bowden, A., Cuellar, A. A., Dronov, S.,
Gilles, E. D., Ginkel, M., Gor, V., Goryanin, I. I., Hed-
ley, W. J., Hodgman, T. C., Hofmeyr, J.-H., Hunter,
P. J., Juty, N. S., Kasberger, J. L., Kremling, A., Kum-
mer, U., Le Nov
`
ere, N., Loew, L. M., Lucio, D.,
Mendes, P., Minch, E., Mjolsness, E. D., Nakayama,
Y., Nelson, M. R., Nielsen, P. F., Sakurada, T., Schaff,
J. C., Shapiro, B. E., Shimizu, T. S., Spence, H. D.,
Stelling, J., Takahashi, K., Tomita, M., Wagner, J.,
Wang, J., and the rest of the SBML Forum (2003). The
systems biology markup language (SBML): a medium
for representation and exchange of biochemical net-
work models. Bioinformatics, 19(4):524–531.
Kanehisa, M. (2002). The kegg database. In ‘In
silico’simulation of biological processes: Novartis
Foundation Symposium 247, volume 247, pages 91–
103. Wiley Online Library.
Kitano, H. (2002). Systems biology: a brief overview. sci-
ence, 295(5560):1662–1664.
Malik-Sheriff, R. S., Glont, M., Nguyen, T. V. N., Tiwari,
K., Roberts, M. G., Xavier, A., Vu, M. T., Men, J.,
Maire, M., Kananathan, S., Fairbanks, E. L., Meyer,
J. P., Arankalle, C., Varusai, T. M., Knight-Schrijver,
V., Li, L., Due
˜
nas-Roca, C., Dass, G., Keating, S. M.,
Park, Y. M., Buso, N., Rodriguez, N., Hucka, M., and
Hermjakob, H. (2020). BioModels — 15 years of
sharing computational models in life science. Nucleic
Acids Research, 48(D1):D407–D415. gkz1055.
Milacic, M., Beavers, D., Conley, P., Gong, C., Gillespie,
M., Griss, J., Haw, R., Jassal, B., Matthews, L., May,
B., et al. (2024). The reactome pathway knowledge-
base 2024. Nucleic Acids Research, 52(D1):D672–
D678.
Picart-Armada, S., Barrett, S. J., Will
´
e, D. R., Perera-
Lluna, A., Gutteridge, A., and Dessailly, B. H. (2019).
Benchmarking network propagation methods for dis-
ease gene identification. PLoS Computational Biol-
ogy, 15(9):e1007276.
Santolini, M. and Barab
´
asi, A.-L. (2018). Predicting per-
turbation patterns from the topology of biological net-
works. PNAS, 115(27):E6375–E6383.
Welsh, C., Xu, J., Smith, L., K
¨
onig, M., Choi, K., and
Sauro, H. M. (2022). libRoadRunner 2.0: a high
performance SBML simulation and analysis library.
Bioinformatics, 39(1):btac770.
Yordanov, P. et al. (2020). Bioswitch: a tool for the de-
tection of bistability and multi-steady state behaviour
in signalling and gene regulatory networks. Bioinfor-
matics, 36(5):1640–1641.
Towards a New Method for Perturbation Analysis in Biochemical Pathways Based on Network Propagation
691
